1. Introduction
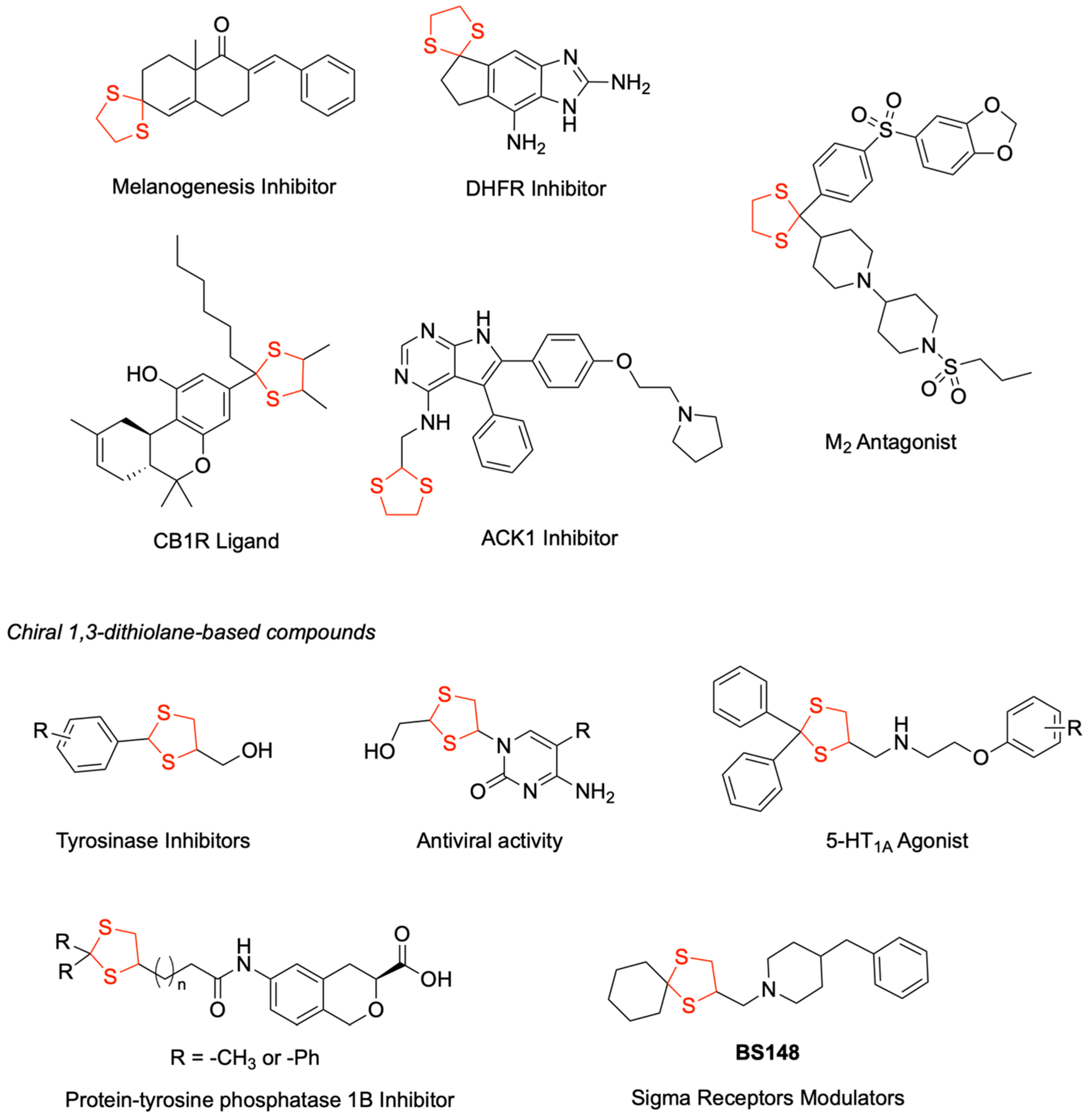
The 1,3-dithiolane heterocycle has been rehabilitated as a chemical scaffold in drug design, being incorporated into diverse therapeutic agents for treating different diseases, including neoplastic, infectious, inflammatory, and neurodegenerative disorders. As depicted in
Figure 1, this scaffold has been the basis of many promising drug candidates. As an example, several M
2 receptor antagonists with enhanced metabolic stability were reported [
1]. Structure–activity relationship studies led to the identification of efficacious melanogenesis inhibitors [
2]. Further medicinal chemistry efforts yielded cannabinoid CB1 receptor ligands possessing favorable drug-like properties and enhanced pharmacodynamic profiles [
3]. The versatility of this scaffold is further demonstrated by its incorporation into dihydrofolate reductase (DHFR) inhibitors, which established a promising foundation for the development of novel anti-infective agents [
4]. Of particular significance, a 1,3-dithiolane-functionalized 4,5,6-trisubstituted-7
H-pyrrolo[2,3-d]pyrimidin-4-amine was identified as a potent ACK1 (Activated Cdc42-associated tyrosine Kinase 1) inhibitor, demonstrating excellent cellular activity, high kinase selectivity, and favorable in vitro metabolic profile. However, suboptimal pharmacokinetic parameters precluded its evaluation in tumor xenograft models [
5].
The incorporation of the 1,3-dithiolane ring, particularly when substituted at position 4, might result in the introduction of a stereogenic center. Several bioactive compounds featuring chiral dithiolane moieties have demonstrated therapeutic potential (
Figure 1). Notable examples include the series of (2-substituted phenyl-1,3-dithiolan-4-yl) methanols that were developed as tyrosinase inhibitors with antimelanogenic effects [
6]. A series of isochroman mono-carboxylic acid derivatives were reported as protein tyrosine phosphatase 1B (PTP1B) inhibitors, where high potency was associated when a dithiolane ring was introduced as a side chain to the isochroman ring [
7]. The therapeutic versatility of 1,3-dithiolane scaffolds was further exemplified by patented nucleoside analogs, which demonstrated promising antiviral activity [
8].
A significant contribution was made by rational drug design when the bioisosteric replacement of 1,3-dithiolane in α1-adrenoceptor antagonists yielded selective 5-HT
1A agonists with enhanced potency. Extending this strategy, 1,4-dioxaspiro[4.5]decane derivatives were successfully transformed into cyclohexylspiro 1,3-dithiolane analogs, which exhibited nanomolar activity as sigma receptor modulators. Particularly noteworthy was compound
BS148 [
9] (
Figure 1), which showed selective toxicity against SK-MEL-2, a metastatic malignant melanoma cell line, while not affecting normal human melanocytes. Furthermore, in PDX models,
BS148 is effective in reducing cell viability and migration properties, showing potential as a future preclinical candidate for treating metastatic melanoma [
10].
Despite the relevance of the 1,3-dithiolane moiety in medicinal chemistry—and the known concept that stereochemistry is an essential feature of biologically active compounds, since it can influence the pharmacodynamic, as well as the pharmacokinetic, properties and impact the toxicology [
11,
12]—the enantiomeric resolution of compounds possessing a stereocenter on this heterocycle has been poorly investigated [
13,
14]. At present, these chiral 1,3-dithiolanes have been synthesized and biologically evaluated as racemates.
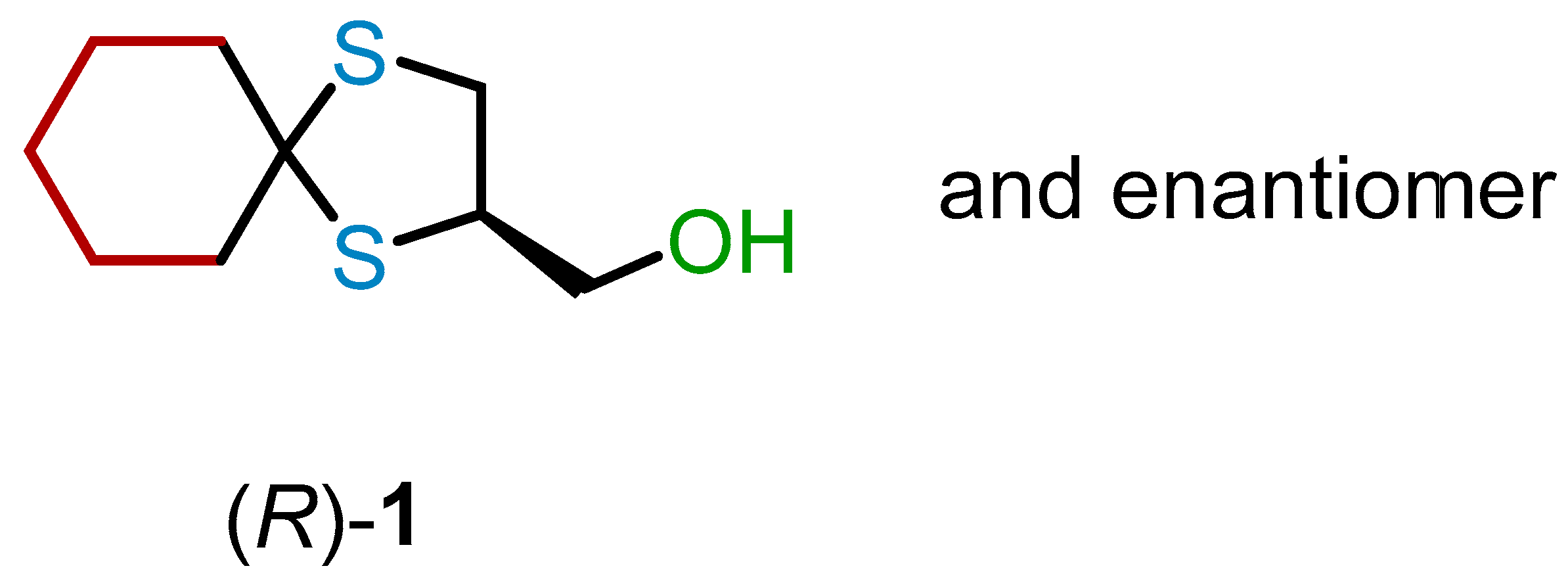
In this work, motivated by our interest in 1,3-dithiolane
1 (
Figure 2), we systematically investigated various approaches for its chiral resolution and the assignment of the absolute configuration (AC) to the enantiomers.
2. Results and Discussion
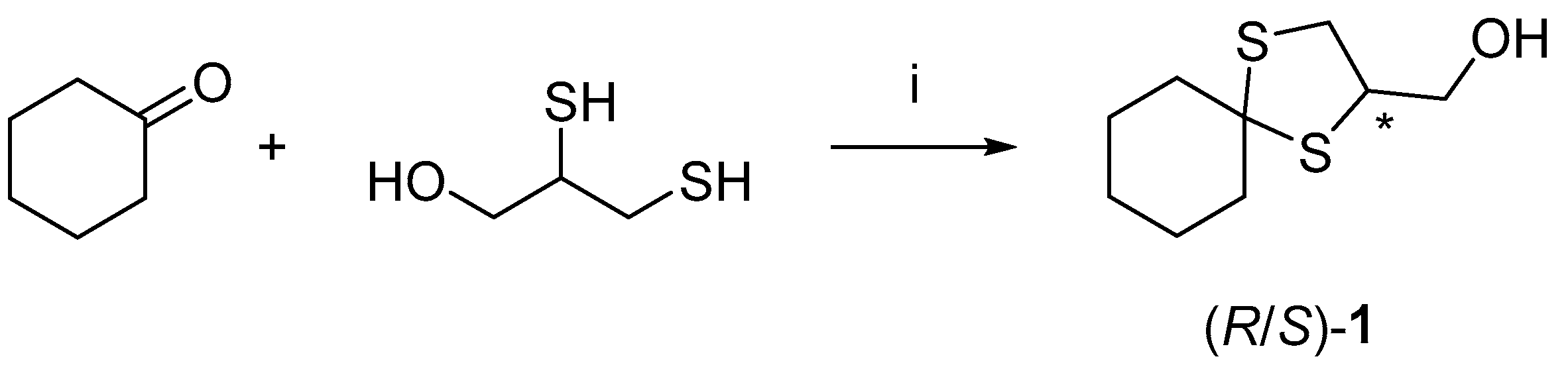
Alcohol (±)-
1 was synthesized on a gram scale using the procedure already published by Franchini et al. [
9] (
Scheme 1).
Initially, an attempt at chiral resolution of compound
1 was performed via diastereomeric esters, a strategy that is still widely employed in industrial settings due to its reliability and simplicity [
15]. Diastereomeric esters were synthetized, using enantiopure carboxylic acids or sulfonyl chlorides (i.e., Fmoc-
L-phenylalanine, Fmoc-
R-tyrosine, (
S)-(+)-10-camphorsulfonyl chloride, and (1
R)-(+)-3,4 dimethyl aryloxypropionic acid). Different solvent systems were systematically evaluated to achieve separation of the resulting diastereomers based on their solubility differences. However, the recovery was poor and insufficient for separation on a hundred-milligram scale. Therefore, RP-HPLC was investigated as an alternative approach for the separation of the diastereomeric esters of
1 on a (semi)preparative scale, followed by hydrolysis to recover enantiomeric
1. Despite these efforts, enantiomeric
1 was not isolated in a suitable amount for further studies, leading to the discontinuation of this approach.
A biocatalytic approach was therefore investigated as a subsequent attempt to identify an efficient and easy-to-scale chiral resolution method. In fact, biocatalysis is universally recognized as a powerful tool for the efficient preparation of homochiral drugs [
16]. Based on our previous experience, alcohol (
R/S)-
1 was converted to the butyryl ester (
R/
S)-
2 (
Scheme 2), as esterification provides a substrate suitable for enzymatic kinetic resolution, where hydrolytic enzymes can selectively cleave one enantiomer of the ester while leaving the other unchanged, thereby enabling the separation of the two enantiomers through different reaction rates [
17].
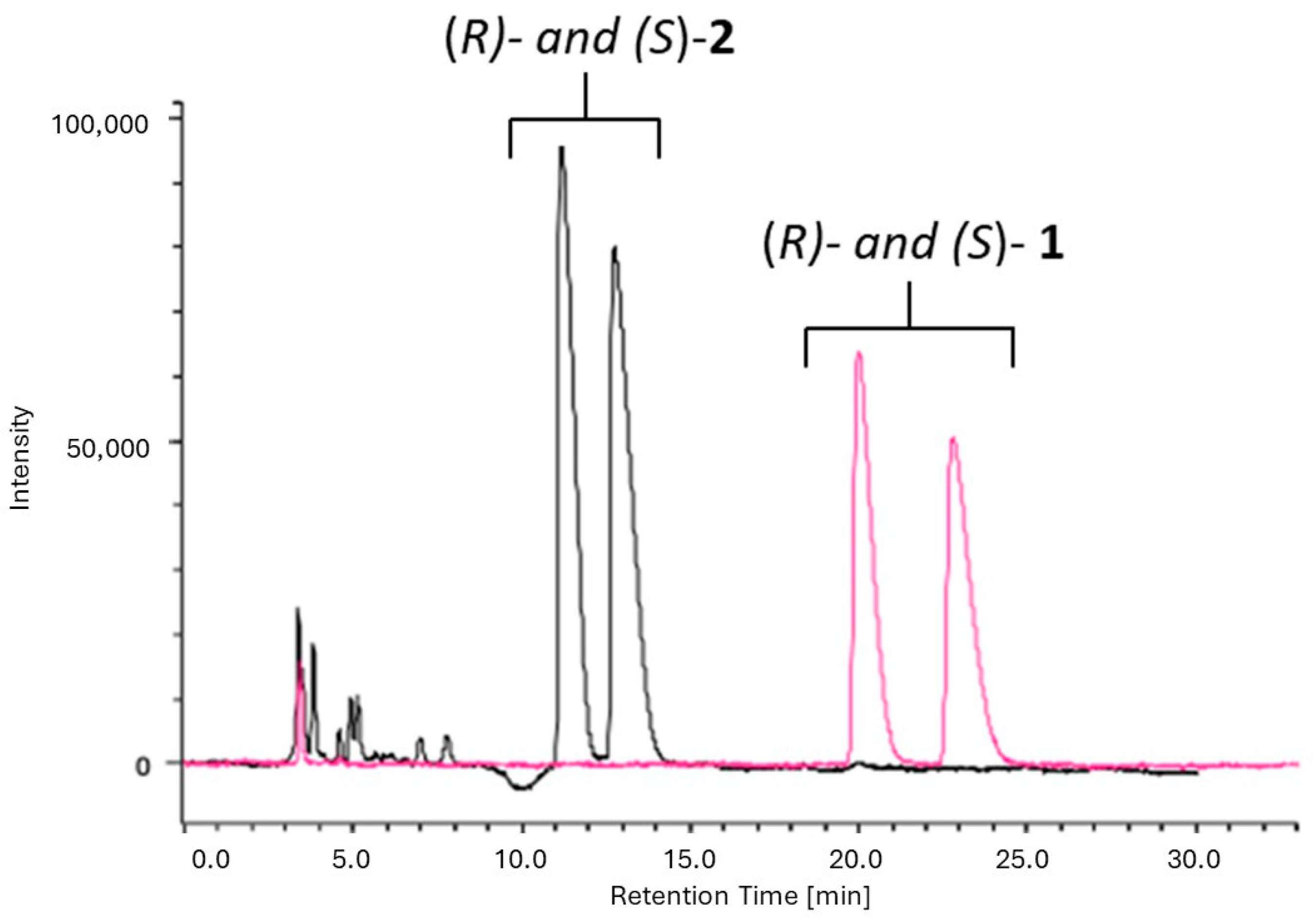
To monitor the enzymatic hydrolysis and assess the enantiomeric purity of product 1, an analytical chromatographic resolution method on Chiral Stationary Phases (CSPs) was developed.
A comparison was performed between two chiral selectors based on polysaccharides consisting of amylose- and cellulose-derived CSPs. Based on our previous experience [
18,
19] Chiralpack IA (amylose-based CSP, 0.46 cm diameter × 25 cm length, 5 μm) and Chiralpak IC (cellulose-based CSP, 0.46 cm diameter × 25 cm length, 5 μm) columns were selected for this study. HPLC screening was performed under two possible chiral separation modes, namely, normal and reverse-phase. Accordingly, the mobile phases investigated were a mixture of
n-hexane (
n-Hex) and 2-propanol (IPA) or ethanol or methyl tert-butyl ether (MTBE) as polar modifiers, or pure alcohols (IPA or ethanol). Several types of mobile phase compositions were investigated by changing the percentages of the mixture components. The main results of the several assessed protocols, expressed in terms of the retention factors (
k1 and
k2), separation factor (α), and resolution factor (
Rs), are reported in
Table S4. Notably, the best chromatographic conditions for separation of the enantiomers of alcohol
1 (see
Table S4, conditions E and H) were also tested for ester
2. Only condition H allowed for the resolution of ester
2, thus enabling the monitoring of the enzymatic reaction progression in one single run. According to the screening results, suitable baseline separation of both alcohol
1 (
k1 = 4.60;
k2 = 5.34; α = 1.16;
Rs = 1.39) and ester
2 (
k1 = 2.18;
k2 = 2.63; α = 1.21;
Rs = 1.60) was obtained on Chiralpak IA with
n-Hex:MTBE 4:1
v/
v at a flow rate of 1 mL/min (
Figure 3).
With the analytical method being established, a set of hydrolytic enzymes, including lipases, esterases, proteases, and acylases, that were previously immobilized in-house (
Figures S1–S3 and Tables S1–S3) on various carriers (except Novozyme435
®, which is commercially available) (
Table 1) were tested [
20,
21]. The initial screening was performed using 10 mM of (
R/S)-
2 in
tert-butanol and water (90:10
v/
v), and 68 units of enzyme
per mg of substrate were incubating at room temperature on a rolling shaker (Scheme S1). The progression of the reaction was monitored by HPLC analysis at two time points (6 and 24 h) to determine the degree of conversion (ester–alcohol) and the enantiomeric excess (
ee) of both ester
2 and alcohol
1. Experimental details about the enzyme immobilization, activity determination, biocatalyzed reaction, and monitoring are reported in the
Supplementary Materials.
All the enzymes tested were able to hydrolyze the substrate (
R/S)-
2 with good conversion rates (>60%). Despite the good conversion, all the lipases tested completely lacked enantioselectivity. Only acylase and ANE yielded the target alcohol
1 with nearly full conversion, although with low enantioselectivity (
ee of 25% and 30% after 24 h, respectively). Subsequent optimization attempts were performed with both acylase and ANE, monitoring the reaction before the 24 h end point (2 h, 4 h, 7 h, 10 h, 13 h, 16 h, 19 h, and 24 h). However, these experiments did not lead to a significant improvement in enantioselectivity (
Figures S4 and S5). As a result, these data limit the applicability of the enzymatic approach on a preparative scale.
We therefore attempted the resolution of (
R/
S)-
1 via (semi)preparative chiral chromatography separation, scaling up the above-described analytical protocol. The chromatographic resolution was carried out on a Chiralpak IA column (250 mm × 1 cm, 5 µm), using a mixture of
n-Hex and MTBE (4:1,
v/
v) at a 5 mL/min flow rate. This method has an essential feature for economic and productive preparative enantiomer separation, i.e., the high solubility of the racemate and enantiomers in the eluent/injection solvent. Conversely, the composition of the mobile phase is a weakness, as it cannot be reused. To overcome the limitation related to the non-recyclable nature of the mobile phase and to improve the environmental sustainability of the protocol, continuous injections of the racemate were carried out. Moreover, injection parameters, such as sample concentrations (10, 20, and 30 mg/mL) and injection volumes (100 and 250 μL), were optimized to achieve a proper balance between the amount of sample injected and the chromatographic resolution. Optimal separation conditions were achieved with cycles of 11 min of continuous sample injections (100 µL) at a 30 mg/mL concentration (
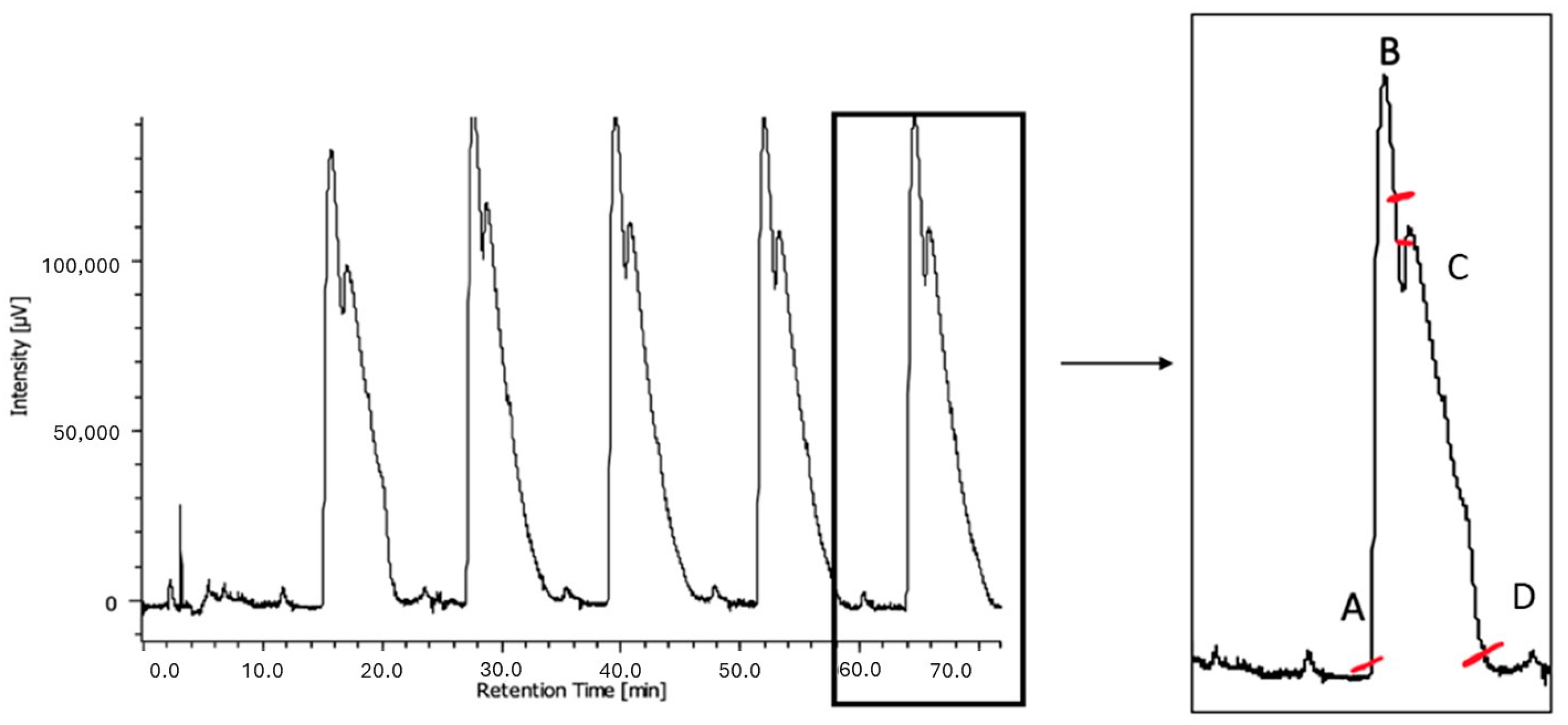
Figure 4).
Through this optimized protocol, 100 mg of racemic
1 was processed in three cycles of injections. The eluate was properly partitioned according to the UV profile (
Figure 4, right), leading to the isolation of 34 mg of the first eluted enantiomer with an
ee of 99.9% (
= −20.5, c ≅ 0.5%, DCM) and 43 mg of the second eluted enantiomer with an
ee of 85.1%. A subsequent purification step of the second fraction (five injections) led to the isolation of 32 mg of the second eluted enantiomer (recovery 64%) with an
ee of 95.6% (
= +17.9; c ≅ 0.5%; DCM).
Summarizing, using the conditions described above, the separation of 100 mg of (R/S)-1 was accomplished within one working day, consuming approximately 3 L of mobile phase.
The quantity of isolated enantiomers of
1 was sufficient for AC assignment studies. Among the experimental methods available, electronic and vibrational circular dichroism (ECD and VCD, respectively) were not applicable due to the lack of reference compounds in the literature to which the VCD or ECD spectra could be compared. Therefore, we opted for X-ray diffraction, which is the gold-standard technique for AC assignment, due to its reliability of data interpretation. Despite different crystallization trials with (−)-
1 being performed using various solvents and techniques such as slow cooling, evaporation, steam, and vapor diffusion [
22], all attempts failed, probably owing to the flexibility of the molecule.
For this reason, we shifted our attention to
BS148 (
Figure 1), which possesses the same spyro cyclohexyldithiolane scaffold and is prepared from
1, based on the hypothesis that it might be more easily crystallized from its precursor owing to its structural features [
9]. Accordingly, (+)-
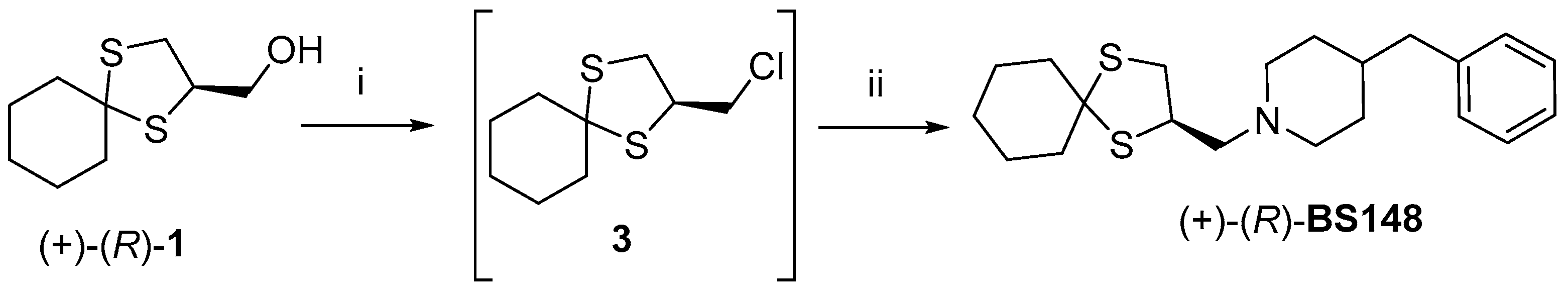
1 was used for the enantioselective synthesis of the corresponding homochiral
BS148. Briefly, as reported in
Scheme 3, (+)-
1 was converted first into the alkyl chloride
3 by reaction with thionyl chloride in anhydrous DCM at room temperature for 15 min, using DMF as a catalyst. Intermediate
3 was reacted with 4-benzylpiperidine under modified S
N2 conditions (the inorganic base was avoided to prevent and/or limit racemization) to achieve
BS148.
To verify whether racemization occurred throughout the synthetic procedure, a HPLC analytical resolution method for
BS148 was set up by making a slight modification to the mobile phase composition that was used for the separation of (
R/S)-
1. The chromatographic eluting conditions, together with the UV and ECD traces, are reported in
Figure 5. The use of the chiroptical detector allowed us to simultaneously determine the enantiomer elution order at the analytical stage.
The analysis showed that the thus obtained BS148 has an ee value of 99.9% ( = +24.8, c ≅ 0.3%, DCM), meaning that, as foreseen, no evidence of racemization in the synthetic steps occurred.
The enantiopure sample of (+)-BS148 was therefore amenable and subjected to crystallization studies.
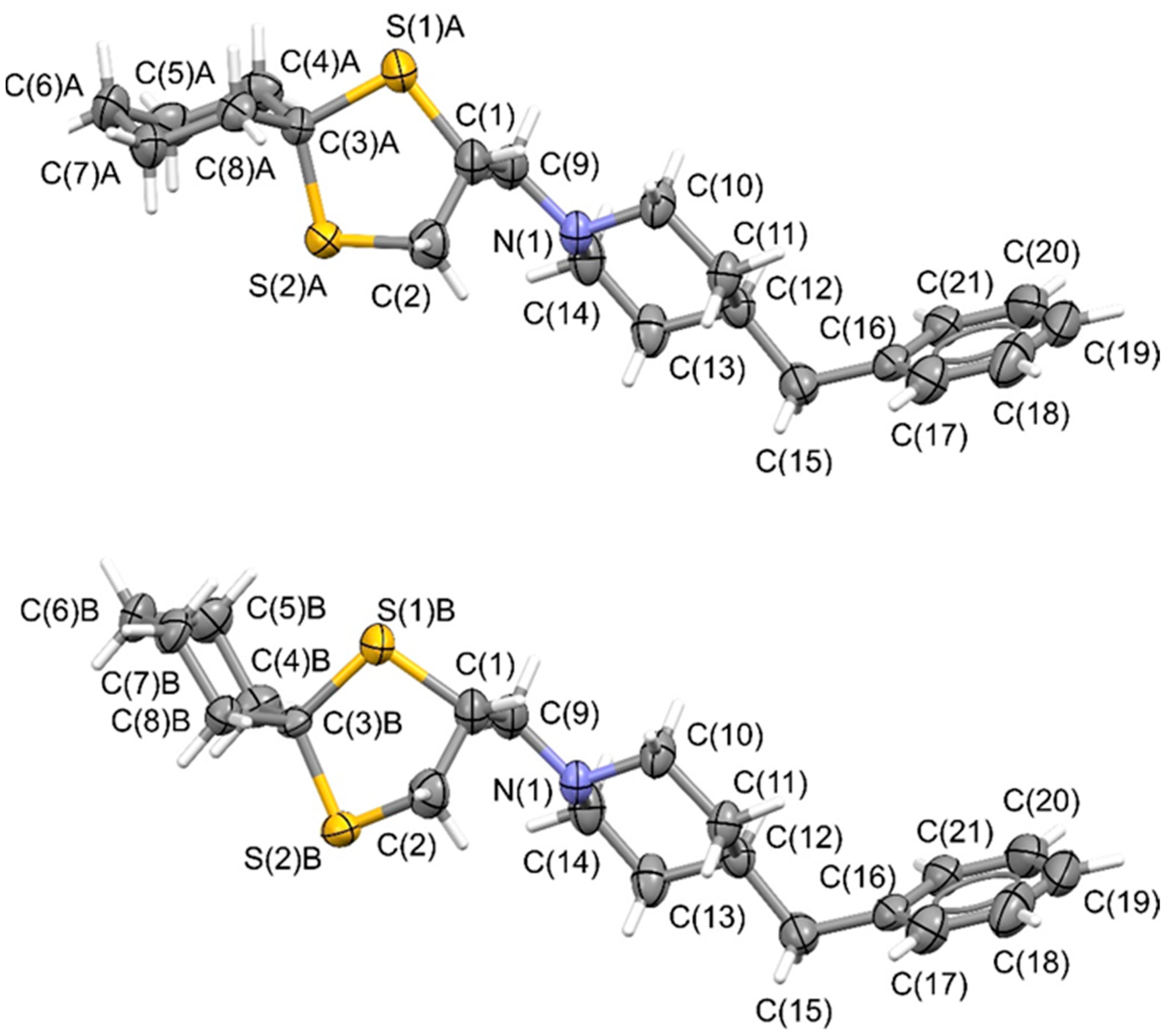
A high-quality needle-shaped single crystal of (+)-
BS148 was successfully obtained through slow-evaporation crystallization from ethanol at room temperature, and a crystallographic analysis was then performed. The crystal structure was solved in the Sohncke
P2
12
12
1 space groups (crystal data are reported in
Table S5) and showed positional disordering resulting from the presence of two mutually exclusive conformations of the cyclohexylspyrodithiolane scaffold (top and bottom of
Figure 6). In these two conformations, the 1,3-dithiolane rings exhibited slightly different spatial orientations, while the cyclohexyl ring was arranged in chair conformations as an almost regular envelope (top of
Figure 6) or in a slightly twisted shape (bottom of
Figure 6). Nevertheless, the chiral C(1) atom was not affected by this positional disorder. To assess the correctness of the determined absolute structure, and therefore the absolute configuration of the chiral C(1) atoms, we investigated the anomalous scattering effects in the measured reflections by analyzing the value of the
x Flack parameter and of its standard uncertainty
u. Based on the anomalous scattering effects [
23], the final
x(
u) Flack parameter was −0.01(7). The
x value of −0.01 was near to 0 (i.e., the value for a correctly assigned absolute structure) but was outside its physically meaningful range (from 0 to 1). Moreover, the
u value of 0.07 was in the range of 0.04–0.10, for which validation of the enantiopurity of the starting material has to be assessed prior to the assignation of the absolute configuration of an analyte [
24]. In our case, the
u value was acceptable for the absolute configuration assignment, since we demonstrated the enantiopurity of the compound that was subjected to the crystallographic studies (
ee > 99.9%, as reported in
Figure 5) [
25,
26,
27].
Accordingly, the combination of the X-ray crystallography and the prior determination and chiroptical characterization by CSP HPLC and in-line CD spectra of the enantiopurity of (+)-BS148 allowed us to unequivocally establish the (R)-configuration for (+)-BS148 and therefore for the key precursor (+)-1.
Summarizing, we developed a (semi)preparative method that is suitable for achieving quick access to the desired enantiomers with high enantiomeric excess and sufficient amounts for subsequent AC studies. Thanks to the high-concentration continuous injections, this approach is efficient in terms of time and solvent consumption. This method may be further developed for production of gram to Kg amounts of pure enantiomers that are needed for studying the role of chirality in biologically active compounds. Moreover, the X-ray analysis data, together with the prior characterization of the enantiopurity of the synthesized (+)-BS148, allowed for the indirect assignment of the AC to its precursor (+)-(R)-1 and to the corresponding enantiomer (−)-(S)-1.
3. Materials and Methods
3.1. General Information
All commercially available reagents and solvents (Merck, Milan, Italy) were reagent-grade and were used without further purification, unless otherwise specified. Air/water-sensitive reactions were performed using oven-dried glassware and under a nitrogen atmosphere. The following solvents and reagents have been abbreviated: ethyl ether (Et2O), dimethyl sulfoxide (DMSO), ethyl acetate (EtOAc), dichloromethane (DCM), ethanol (EtOH), dimethylformamide (DMF), 4-dimethylaminopyridine (DMAP), n-hexane (n-Hex), n-propanol (IPA), and methyl tert-butyl ether (MTBE). All the compounds and intermediates were purified by flash chromatography on a silica gel stationary phase (60 Å; 230–400 Mesh). Reactions were monitored by thin-layer chromatography (TLC) on silica gel pre-coated glass-backed plates (60-F254; Merk) and visualized using UV light (λ = 254 and 366 nm, MinUVIS DESAGA® Sarstedt-GRUPPE, Wiesloch, Germany), ninhydrin, or a cerium ammonium sulfate molybdate solution. NMR spectra were recorded on a Bruker Avance 400 spectrometer equipped with a BBI 5 mm probe, with 1H at 400.134 MHz and 13C at 100.62 MHz. Spectra were processed with the Top Spin software 4.4.0 from Bruker. Proton and carbon chemical shifts were referenced to the residual solvent peak. Chemical shifts are expressed in parts per million (ppm or δ). Coupling constants are reported in Hz. Multiplicity patterns are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and brs (broad signal). Enzymatic activity assays were monitored by Titrator 718 stat (pH-Stat) Tritino from Metrohm (Herisau, Switzerland). HPLC-UV/Vis analysis was carried out on a JASCO (Tokyo, Japan) system composed of a PU-2089 pump, AS-2055 autosampler, CO-4065 column oven, MD-1510 photodiode array (PDA) detector, and CD-2095 Electronic Circular Dichroism (ECD) Plus detector. The (semi)preparative chromatographic resolutions were performed on a Jasco system (JASCO Europe, Cremella, LC, Italy) equipped with a PU-2089 plus quaternary gradient pump, AS-2055 plus autosampler, MD-1510 Photo Diode Array (PDA) detector, and Electronic Circular Dichroism (ECD) 2095 Plus detector. Chromatograms were acquired and processed using the ChromNAV v 2.0 software (Tokyo, Japan). Optical rotation values were measured on a JASCO photoelectric polarimeter DIP 1000 with a 0.5 dm quartz cell at the sodium D line (λ = 589 nm); compounds were dissolved in DCM at a concentration of 0.5% (w/v), unless otherwise stated. X-ray diffraction data were collected on a Enraf-Nonius CAD4 four-circle diffractometer (Enraf-Nonius, Delft, The Netherlands) at room temperature with graphite monochromated Mo-Kα X-radiation (λ = 0.7107 Å).
3.2. HPLC Analysis
Enantiomeric resolutions for (R/S)-1, (R/S)-2, (R/S)-BS148, and (+)-(R)-BS148 were achieved as follows:
Method A was adopted for (R/S)-1 and (R/S)-2. Chiralpak IA column (4.6 × 250 mm, 5 µm) was used with n-Hex/MTBE 4:1 (v/v) as the mobile phase, with a flow rate of 1 mL/min, a sample concentration of 1 mg/mL, an injection volume of 10 μL, and UV/vis-HPLC trace (λ = 240 nm). The elution allowed us to obtain a baseline separation of both compounds. Results are expressed as retention factors (k1 and k2), separation (α), and resolution (RS) factors. Specifically, (R/S)-1 showed k1 = 4.60; k2 = 5.34; α = 1.16; and Rs = 1.39. The ester (R/S)-2 showed k1 = 2.18; k2 = 2.63; α= 1.21; and Rs = 1.60.
Method B was adopted for BS148. A Chiralpak IA column (4.6 × 250 mm, 5 µm) was used with n-Hex/MTBE/Diethylamine 85:15:0.1 (v/v/v) as the mobile phase, with a flow rate of 1 mL/min, a sample concentration of 1 mg/mL, an injection volume of 10 μL, UV/vis-HPLC trace (λ = 240 nm), and ECD-HPLC trace (λ = 240 nm). The elution allowed us to obtain a baseline separation. Results are expressed as retention factors (k1 and k2), separation (α), and resolution (RS) factors. Specifically, (R/S)-BS148 showed k1 = 0.68; k2 = 1.00; α = 1.42; and RS = 2.20. (+)-(R)-BS148 showed k1 = 0.68.
3.3. Synthesis of (1,4-Dithiaspiro[4.5]decan-2-yl)methanol (R/S)-1
Under a nitrogen atmosphere and at room temperature, 2,3-dimercaptopropan-1-ol (1.26 g, 10.19 mmol, 1 equiv.) and HClO
4 absorbed on SiO
2 (70%
v/
w) (20 mg
per mmol) were added to cyclohexanone (1 g, 10.19 mmol, 1 equiv.) The suspension was reacted neat under the same conditions for 1 h and quenched by the addition of EtOAc. The suspension was filtered and concentrated. The crude product was directly purified over a silica gel (ratio crude/silica 1:100, eluent DCM 100%) to obtain the desired product (310 mg, 30.5% yield) as a whitish oil. The
1H and
13C spectra of the racemate are in accordance with the literature [
17].
3.4. Synthesis of (1,4-Dithiaspiro[4.5]decan-2-yl)methyl Butyrate, (R/S)-2
(±)-1 (250 mg, 1.22 mmol, 1 equiv.) was dissolved in 7 mL of anhydrous DCM at room temperature and under a nitrogen atmosphere, followed by the addition of butyryl chloride (252 μL, 1.22 mmol, 1 equiv.), triethylamine (278.3 μL, 2.447 mmol, 2 equiv.), and a catalytic amount of DMAP. After 8 h, the reaction was stopped. The mixture was concentrated under reduced pressure. The crude product was resuspended in Et2O, and the organic phase was washed with 10% v/v HCl and a saturated solution of NaHCO3. The organic phase was dried over anhydrous Na2SO4 and concentrated to obtain 315 mg (95% yield) of the titled compound as a white oil. 1H NMR (400 MHz, CDCl3): δ 4.18–4.11 (dd, 1H), 4.01–4.04 (dd, 1H), 3.89–3.81 (m, 1H), 3.29–3.22 (dd, 1H), 3.17–3.11 (dd, 1H), 2.26–2.19 (t, 2H), 1.97–1.87 (dt, 4H), 1.66–1.48 (m, 6H), 1.37–1.30 (m, 2H), and 0.92–0.85 (t, 3H).
3.5. (Semi)preparative HPLC Separation
A Hamilton (Reno, NV, USA) syringe (syringe volume: 200 µL; loop: 200 µL) was employed for the (semi)preparative chiral resolutions. (Semi)preparative enantiomer (R/S)-1 separation was performed on a Chiralpak IA (250 mm × 1 cm, 5 µm), using n-Hex/MTBE 4:1 (v/v) as mobile phase with a flow rate of 5 mL/min, a sample concentration of 30 mg/mL, and an injection volume of 100 µL.
The processing of 100 mg of racemic 1 resulted in 34.1 mg of the first eluted enantiomer (ee = 99.95%, recovery 68%) and 43.4 mg of the second eluted enantiomer (ee of 85.1%), together with 17.4 mg of an intermediate fraction as a mixture of the two isomers. A subsequent purification step of the second fraction led to the isolation of 32.1 mg of the second eluted enantiomer (ee of 95.60%, recovery 64%). The chiroptical properties of the enantiomers are reported as follows: (−)-(S)-1: = −20.5 (c ≅ 0.5%, DCM), ee = 99.95%; (+)-(R)-1: = + 17.9 (c ≅ 0.5%, DCM), ee = 95.60%.
3.6. Synthesis of 1-((1,4-Dithiaspiro[4.5]decan-2-yl)methyl)-4-benzylpiperidine (+)-(R)-BS148
To a solution of (±)-1 (34 mg, 0.16 mmol, 1 equiv.) in anhydrous DCM, at room temperature and under a nitrogen atmosphere, thionyl chloride (21.8 µL, 0.32 mmol, 2 equiv.) and a catalytic amount of DMF were added, and the mixture was stirred under the same conditions for 15 min. The reaction was quenched by the addition of Et2O, and the organic phase was washed with a saturated solution of NaHCO3 and brine. The organic phase was dried over anhydrous Na2SO4 and concentrated to obtain the corresponding alkyl chloride 3 (33.5 mg, 94% yield), which was directly used in the next step without further purification.
Chloride 3 (33.5 mg, 0.15 mmol, 1 equiv.) was solubilized in DMSO (2 mL), and 4-benzyl piperidine (52.7 mg, 0.30 mmol, 2 equiv.) and KI (cat.) were added. The mixture was reacted at 90 °C overnight. Thereafter, the reaction was quenched with a saturated solution of Na2CO3, and the aqueous phase was extracted with EtOAc. The organic phase was washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude was purified over a silica gel (ratio crude/silica 1:100, eluent n-Hex/AcOEt 8:2) to obtain 37.9 mg (70% yield) of the titled compound as a pale-yellow oil. TLC Rf: 0.37 (eluent n-Hex/AcOEt 8:2).
1H NMR (400 MHz, Chloroform-d) δ 1.30–1.67 (m, 11H), 1.91–2.11 (m, 6H), 2.46 (dd, J = 6.4, 12.8 Hz, 1H), 2.58 (d, J = 6.8 Hz, 2H), 2.71 (dd, J = 8.4, 12.4 Hz, 1H), 2.89–2.97 (m, 2H), 3.24 (dd, J = 6.4, 12.0 Hz, 1H), 3.33 (dd, J = 4.8, 6.8 Hz, 1H), 3.91–3.94 (m, 1H), 7.18–7.35 (m, 5H). (+)-(R)-BS148: ( = +24.8, c ≅ 0.3%, DCM), and ee = 99.9%.
3.7. X-Ray Crystallography Diffraction Analysis
A single crystal of (+)-
BS148 was obtained through slow-evaporation crystallization from ethanol at room temperature. Data reduction (including intensity integration, background, and Lorentz and polarization corrections) was performed with the WinGX software package—version 2023 [
28], and absorption effects were evaluated by using the psi-scan semi-empirical method [
29]. The absorption effects were found to be negligible, and absorption correction was not applied to the data. The crystal structure was solved by direct methods (SIR 97) [
30] and refined by full-matrix least-square procedures on
F2 using all reflections (SHELXL-2019/3) [
31]. All non-H atoms were refined anisotropically, and hydrogen atoms were placed at calculated positions with the appropriate AFIX instructions and refined using a riding model. Positional disorder affected the 1,3-dithiolane ring and the terminal cyclohexane ring, which adopted two different conformations that were mutually exclusive and statistically distributed. The refined ratio between these two alternative conformations is 0.639(7):0.361(7). Crystal data are reported in
Table S4. CCDC 2333985 contains supplementary crystallographic data for the compound under study. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}