
Understanding the Preclinical Efficacy of Antibody–Drug Conjugates

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Selective Expression of TAAs for Approved ADCs
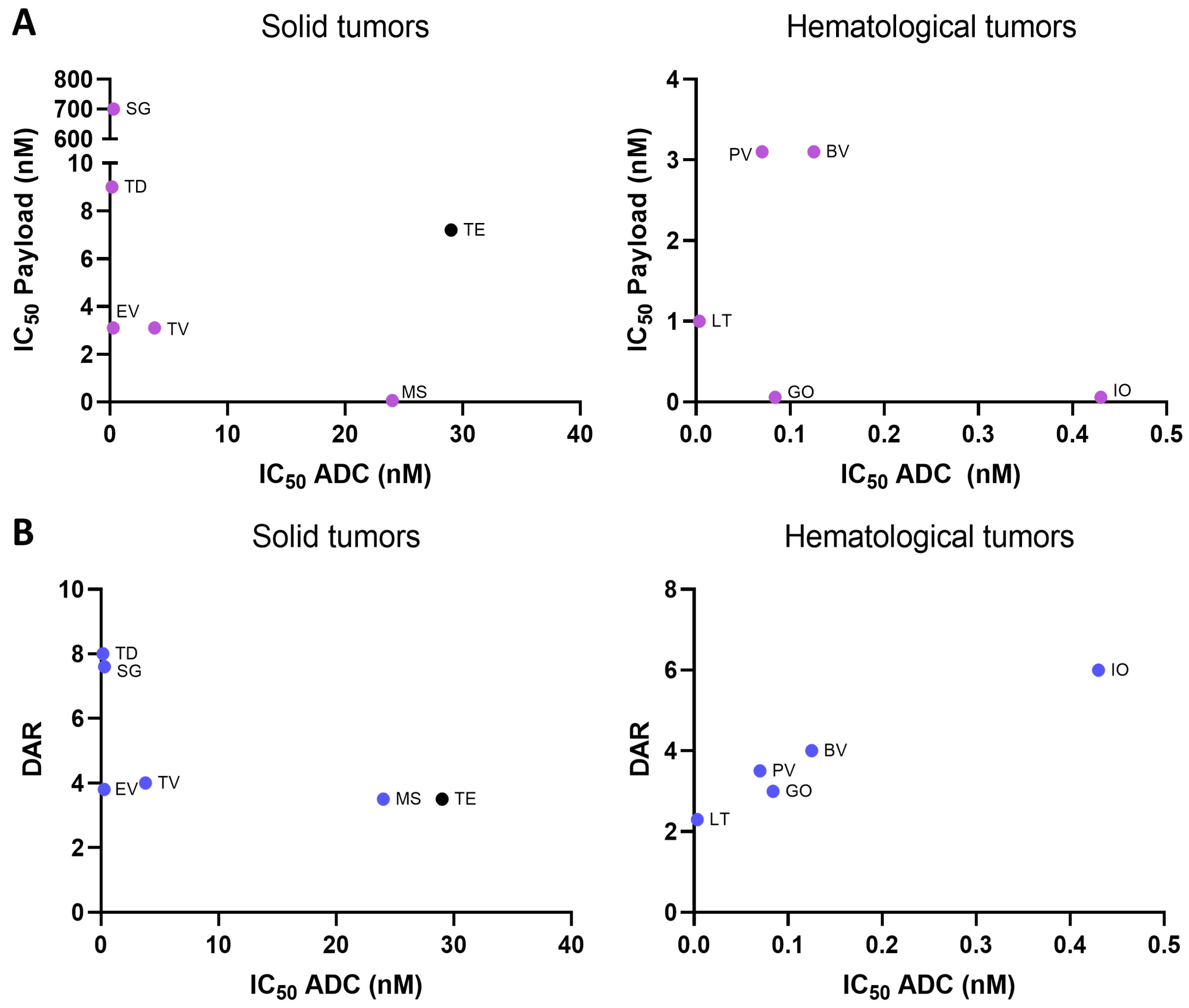
2.2. Payload Potency and Drug-to-Antibody Ratio (DAR) in Relation to Preclinical Activity
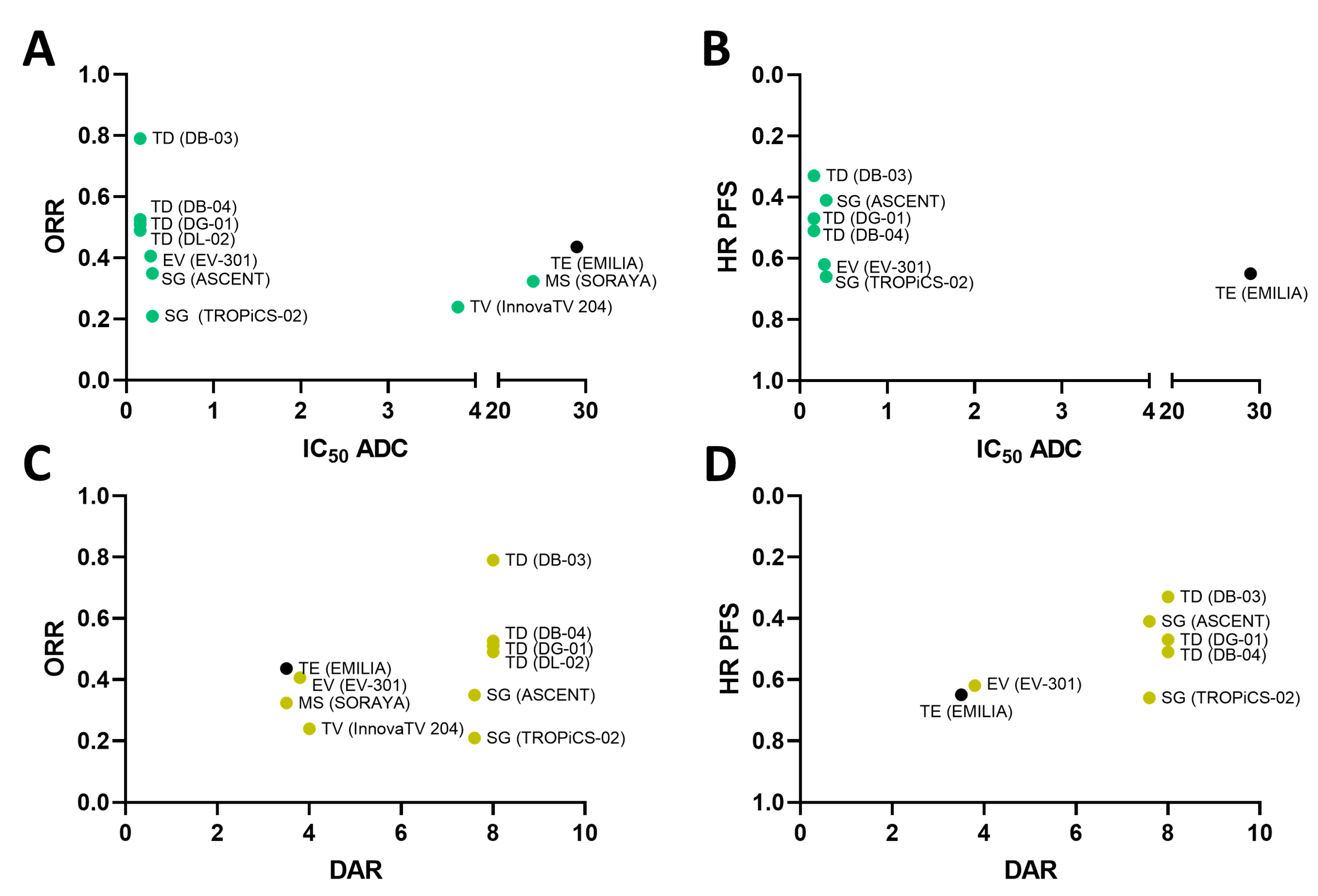
2.3. Payload Potency and Drug-to-Antibody Ratio (DAR) in Relation to Clinical Activity
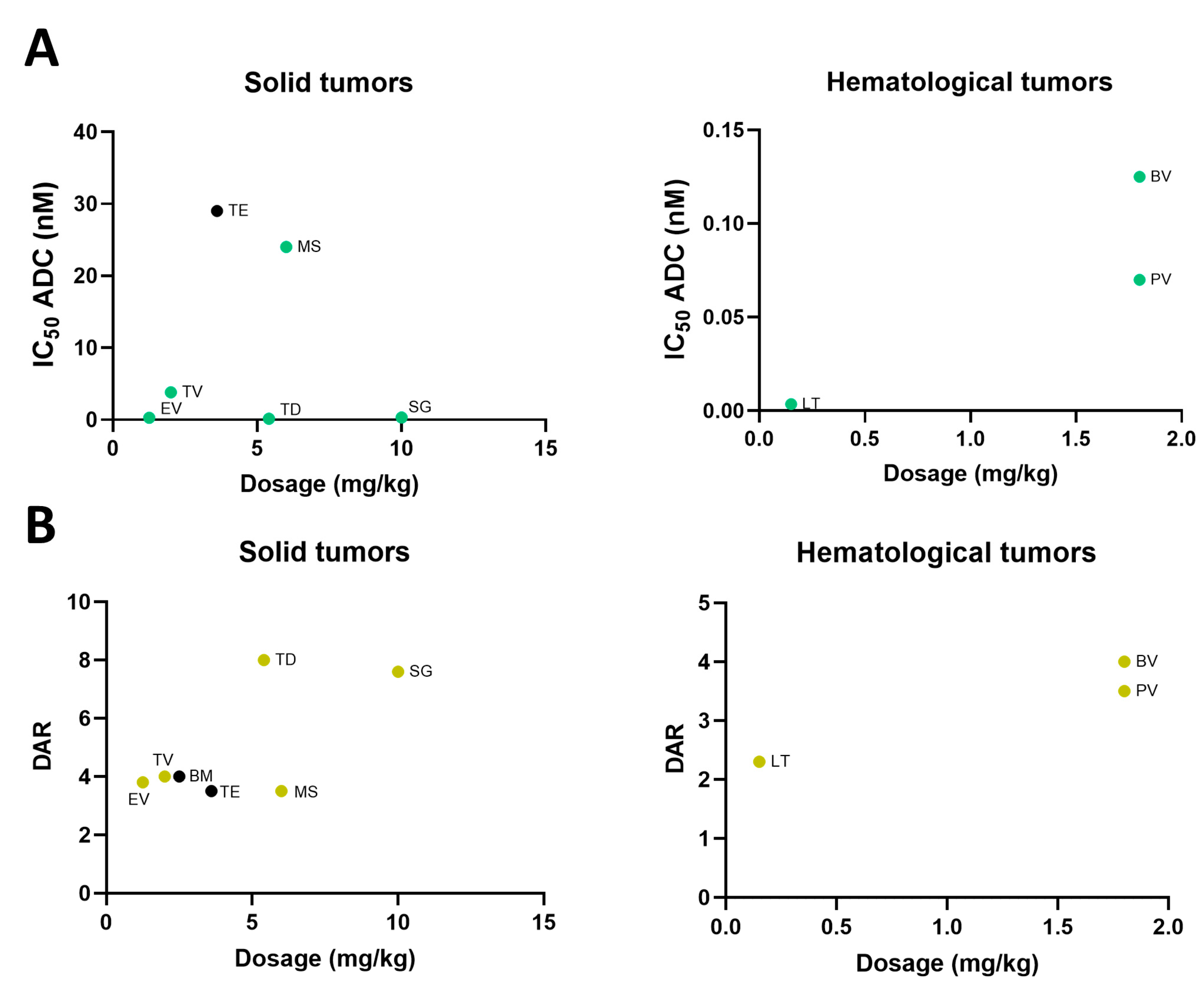
2.4. Dose Selection of Approved ADC
2.5. Evaluation of ADCs with Adequate Physicochemical Properties and Conjugation Type
3. Discussion
4. Materials and Methods
4.1. Data Extraction
4.2. Transcriptomic Extraction and Data Analysis
4.3. Extraction, Collection, and Analysis of Preclinical and Clinical Data from ADCs
4.4. Evaluation of Physicochemical Properties
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | Adrenocortical carcinoma |
| ADCs | Antibody–drug conjugates |
| ADME | Absorption, Distribution, Metabolism, and Excretion |
| BLCA | Bladder Urothelial Carcinoma |
| BRCA | Breast invasive carcinoma |
| BV | Brentuximab vedotin |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangiocarcinoma |
| COAD | Colon adenocarcinoma |
| DAR | drug antibody ratio |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma |
| ESCA | Esophageal carcinoma |
| EV | Enfortumab vedotin |
| GBM | Glioblastoma multiforme |
| GO | Gemtuzumab ozogamicin |
| HNSC | Head and Neck squamous cell carcinoma |
| IO | Inotuzumab ozogamicin |
| KICH | Kidney Chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LAML | Acute Myeloid Leukemia |
| LGG | Brain Lower Grade Glioma |
| LIHC | Liver hepatocellular carcinoma |
| LT | Loncastuximab tesirine |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| MS | Mirvetuximab soravtansine |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCP | Physicochemical properties |
| PCPG | Pheochromocytoma and Paraganglioma |
| PRAD | Prostate adenocarcinoma |
| PV | Polatuzumab vedotin |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SG | Sacituzumab govitecan |
| SKCM | Skin Cutaneous Melanoma |
| SOC | standard of care |
| STAD | Stomach adenocarcinoma |
| TAAs | Tumor-associated antigens |
| TD | Trastuzumab deruxtecan |
| TE | Trastuzumab emtansine |
| TF | tissue factor |
| TGCT | Testicular Germ Cell Tumors |
| THCA | Thyroid carcinoma |
| THYM | Thymoma |
| TPM | transcripts per million |
| TV | Tisotumab vedotin |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UCS | Uterine Carcinosarcoma |
References
- Ocaña, A.; García-Alonso, S.; Amir, E.; Pandiella, A. Refining Early Antitumoral Drug Development. Trends Pharmacol. Sci. 2018, 39, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Settleman, J.; Fernandes Neto, J.M.; Bernards, R. Thinking Differently about Cancer Treatment Regimens. Cancer Discov. 2021, 11, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Amir, E.; Yeung, C.; Seruga, B.; Tannock, I.F. How valid are claims for synergy in published clinical studies? Ann. Oncol. 2012, 23, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Pandiella, A.; Siu, L.L.; Tannock, I.F. Preclinical development of molecular-targeted agents for cancer. Nat. Rev. Clin. Oncol. 2010, 8, 200–209. [Google Scholar] [CrossRef]
- Aloss, K.; Hamar, P. Recent Preclinical and Clinical Progress in Liposomal Doxorubicin. Pharmaceutics 2023, 15, 893. [Google Scholar] [CrossRef] [PubMed]
- Zinn, S.; Vazquez-Lombardi, R.; Zimmermann, C.; Sapra, P.; Jermutus, L.; Christ, D. Advances in antibody-based therapy in oncology. Nat. Cancer 2023, 4, 165–180. [Google Scholar] [CrossRef]
- Tsuchikama, K.; Anami, Y.; Ha, S.Y.Y.; Yamazaki, C.M. Exploring the next generation of antibody-drug conjugates. Nat. Rev. Clin. Oncol. 2024, 21, 203–223. [Google Scholar] [CrossRef]
- Nieto-Jiménez, C. Uncovering therapeutic opportunities in the clinical development of antibody-drug conjugate and immune checkpoint blockade. Clin. Transl. Med. 2023, 13, e1329. [Google Scholar] [CrossRef]
- López de Sá, A.; Díaz-Tejeiro, C.; Poyatos-Racionero, E.; Nieto-Jiménez, C.; Paniagua-Herranz, L.; Sanvicente, A.; Calvo, E.; Pérez-Segura, P.; Moreno, V.; Moris, F.; et al. Considerations for the design of antibody drug conjugates (ADCs) for clinical development: Lessons learned. J. Hematol. Oncol. 2023, 16, 118. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)-A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Luci, C.; García-Alonso, S.; Díaz-Rodríguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocaña, A.; Pandiella, A. Resistance to the Antibody-Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res. 2017, 77, 4639–4651. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, E.; Gandullo-Sánchez, L.; Ocaña, A.; Pandiella, A. Novel ADCs and Strategies to Overcome Resistance to Anti-HER2 ADCs. Cancers 2021, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- O’Brien Laramy, M.N.; Luthra, S.; Brown, M.F.; Bartlett, D.W. Delivering on the promise of protein degraders. Nat. Rev. Drug Discov. 2023, 22, 410–427. [Google Scholar] [CrossRef]
- Juan, A.; Cimas, F.J.; Bravo, I.; Pandiella, A.; Ocaña, A.; Alonso-Moreno, C. Antibody Conjugation of Nanoparticles as Therapeutics for Breast Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6018. [Google Scholar] [CrossRef]
- Niza, E.; Noblejas-lópez, M.D.M.; Bravo, I.; Nieto-jiménez, C.; Castro-osma, J.A.; Canales-vázquez, J.; Lara-Sanchez, A.; Galán Moya, E.M.; Burgos, M.; Ocaña, A.; et al. Trastuzumab-Targeted Biodegradable Nanoparticles for Enhanced Delivery of Dasatinib in HER2+ Metastasic Breast Cancer. Nanomaterials 2019, 9, 1793. [Google Scholar] [CrossRef]
- Niza, E.; Nieto-Jiménez, C.; Noblejas-López, M.D.M.; Bravo, I.; Castro-Osma, J.A.; de la Cruz-Martínez, F.; Martínez de Sarasa Buchaca, M.; Posadas, I.; Canales-Vázquez, J.; Lara-Sanchez, A.; et al. Poly(Cyclohexene Phthalate) Nanoparticles for Controlled Dasatinib Delivery in Breast Cancer Therapy. Nanomaterials 2019, 9, 1208. [Google Scholar] [CrossRef]
- Ocaña, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Hegg, R.; Chung, W.P.; Im, S.A.; Jacot, W.; Ganju, V.; Chiu, J.W.Y.; Xu, B.; Hamilton, E.; Madhusudan, S.; et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: Updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 2023, 401, 105–117. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Lorusso, D.; Oaknin, A.; Pignata, S.; Dean, A.; Denys, H.; Colombo, N.; Van Gorp, T.; Konner, J.A.; Marin, M.R.; et al. Efficacy and Safety of Mirvetuximab Soravtansine in Patients with Platinum-Resistant Ovarian Cancer with High Folate Receptor Alpha Expression: Results from the SORAYA Study. J. Clin. Oncol. 2023, 41, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Hu, X.; Dent, R.A.; Yonemori, K.; Barrios, C.H.; O’Shaughnessy, J.; Wildiers, H.; Zhang, Q.; Im, S.A.; Saura, C.; et al. Trastuzumab deruxtecan (T-DXd) vs physician’s choice of chemotherapy (TPC) in patients (pts) with hormone receptor-positive (HR+), human epidermal growth factor receptor 2 (HER2)-low or HER2-ultralow metastatic breast cancer (mBC) with prior endocrine therapy (ET): Primary results from DESTINY-Breast06 (DB-06). J. Clin. Oncol. 2024, 42 (Suppl. S17), LBA1000. [Google Scholar] [CrossRef]
- Colombo, R.; Rich, J.R. The therapeutic window of antibody drug conjugates: A dogma in need of revision. Cancer Cell 2022, 40, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the safety of antibody-drug conjugates for patients with solid tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Tejeiro, C.; López de Sá, A.; Poyatos-Racionero, E.; Ballestín, P.; Bartolomé, J.; Calvo, E.; Moreno, V.; Moris, F.; Pérez-Segura, P.; Gyorffy, B.; et al. Understanding the Preclinical Efficacy of Antibody–Drug Conjugates. Int. J. Mol. Sci. 2024, 25, 12875. https://doi.org/10.3390/ijms252312875
Díaz-Tejeiro C, López de Sá A, Poyatos-Racionero E, Ballestín P, Bartolomé J, Calvo E, Moreno V, Moris F, Pérez-Segura P, Gyorffy B, et al. Understanding the Preclinical Efficacy of Antibody–Drug Conjugates. International Journal of Molecular Sciences. 2024; 25(23):12875. https://doi.org/10.3390/ijms252312875
Chicago/Turabian StyleDíaz-Tejeiro, Cristina, Alfonso López de Sá, Elisa Poyatos-Racionero, Pablo Ballestín, Jorge Bartolomé, Emiliano Calvo, Víctor Moreno, Francisco Moris, Pedro Pérez-Segura, Balazs Gyorffy, and et al. 2024. "Understanding the Preclinical Efficacy of Antibody–Drug Conjugates" International Journal of Molecular Sciences 25, no. 23: 12875. https://doi.org/10.3390/ijms252312875
APA StyleDíaz-Tejeiro, C., López de Sá, A., Poyatos-Racionero, E., Ballestín, P., Bartolomé, J., Calvo, E., Moreno, V., Moris, F., Pérez-Segura, P., Gyorffy, B., Pandiella, A., & Ocaña, A. (2024). Understanding the Preclinical Efficacy of Antibody–Drug Conjugates. International Journal of Molecular Sciences, 25(23), 12875. https://doi.org/10.3390/ijms252312875