
LRRK2 G2019S Mutated iPSC-Derived Endothelial Cells Exhibit Increased α-Synuclein, Mitochondrial Impairment, and Altered Inflammatory Responses
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Differentiation and Characterization of hiPSC-Derived Endothelial Cells
2.2. LRRK2 Mutation Induces Moderate Changes in Transcriptomics of Endothelial Cells
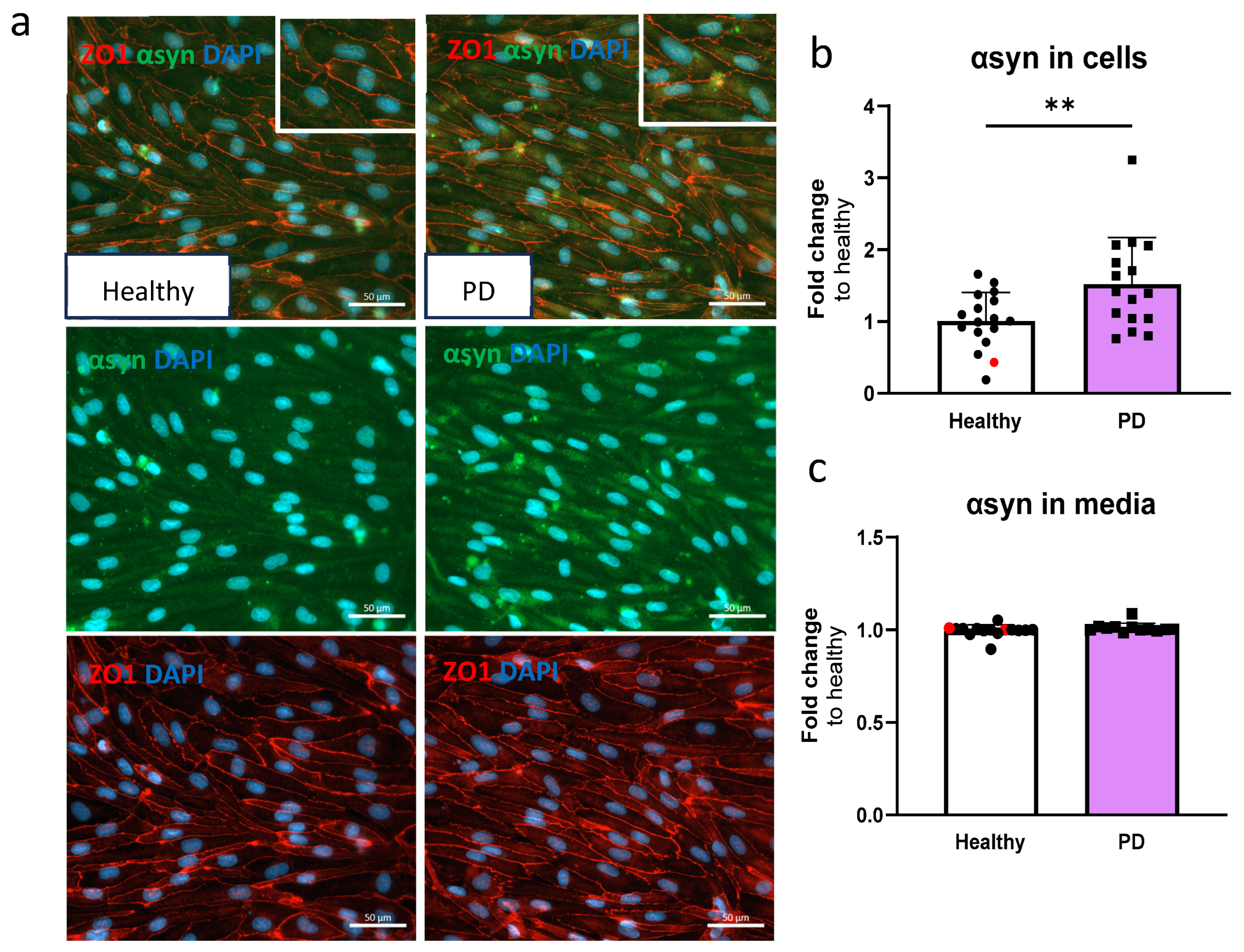
2.3. LRRK2 Mutant Endothelial Cells Have Higher Levels of α-Synuclein
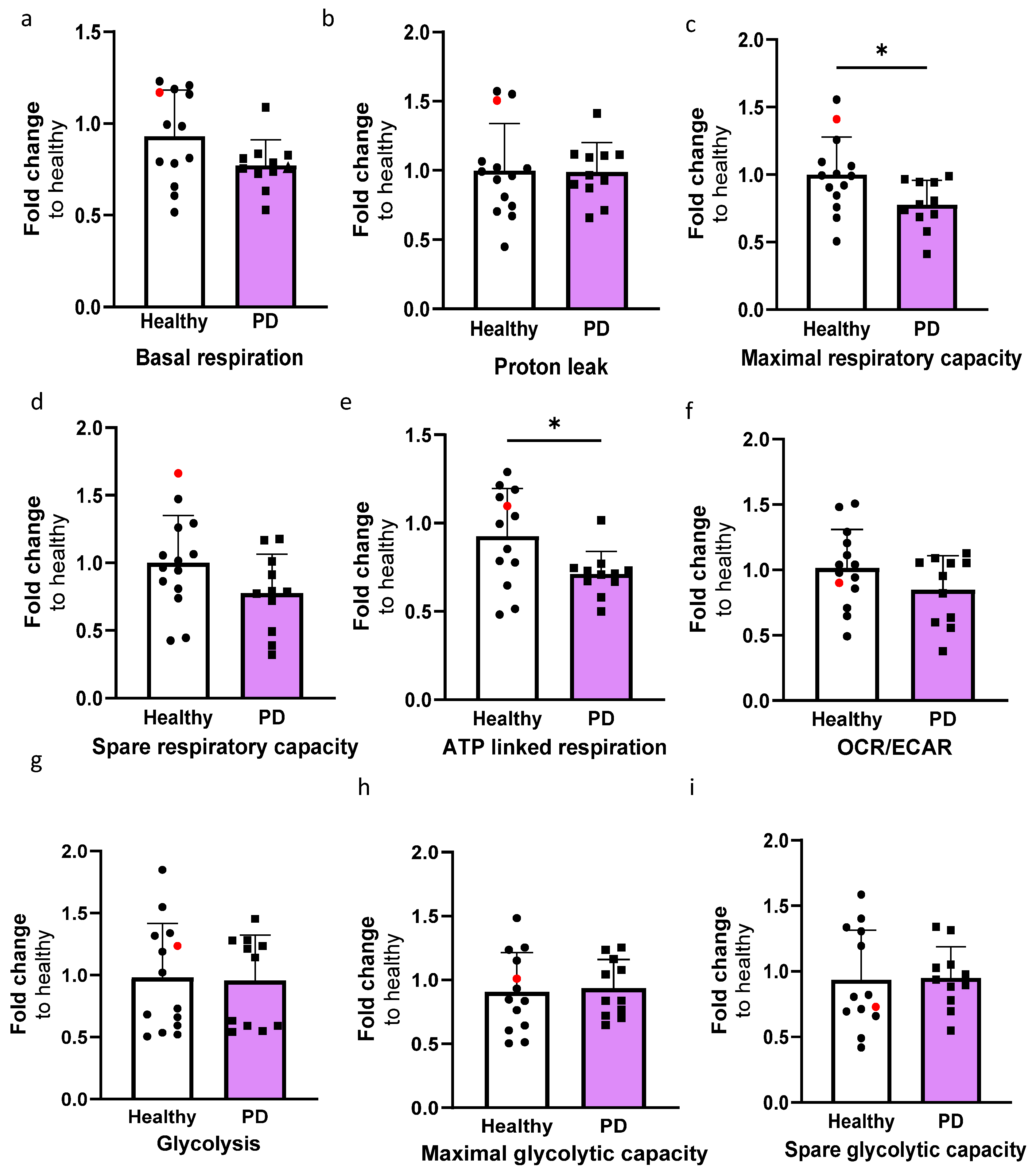
2.4. LRRK2 Mutant Endothelial Cells Have Decreased Maximal and ATP Linked Respiration
2.5. RNA Transcriptomics Reveal More Prominent Effect in PD Endothelial Cells After TNFα Exposure
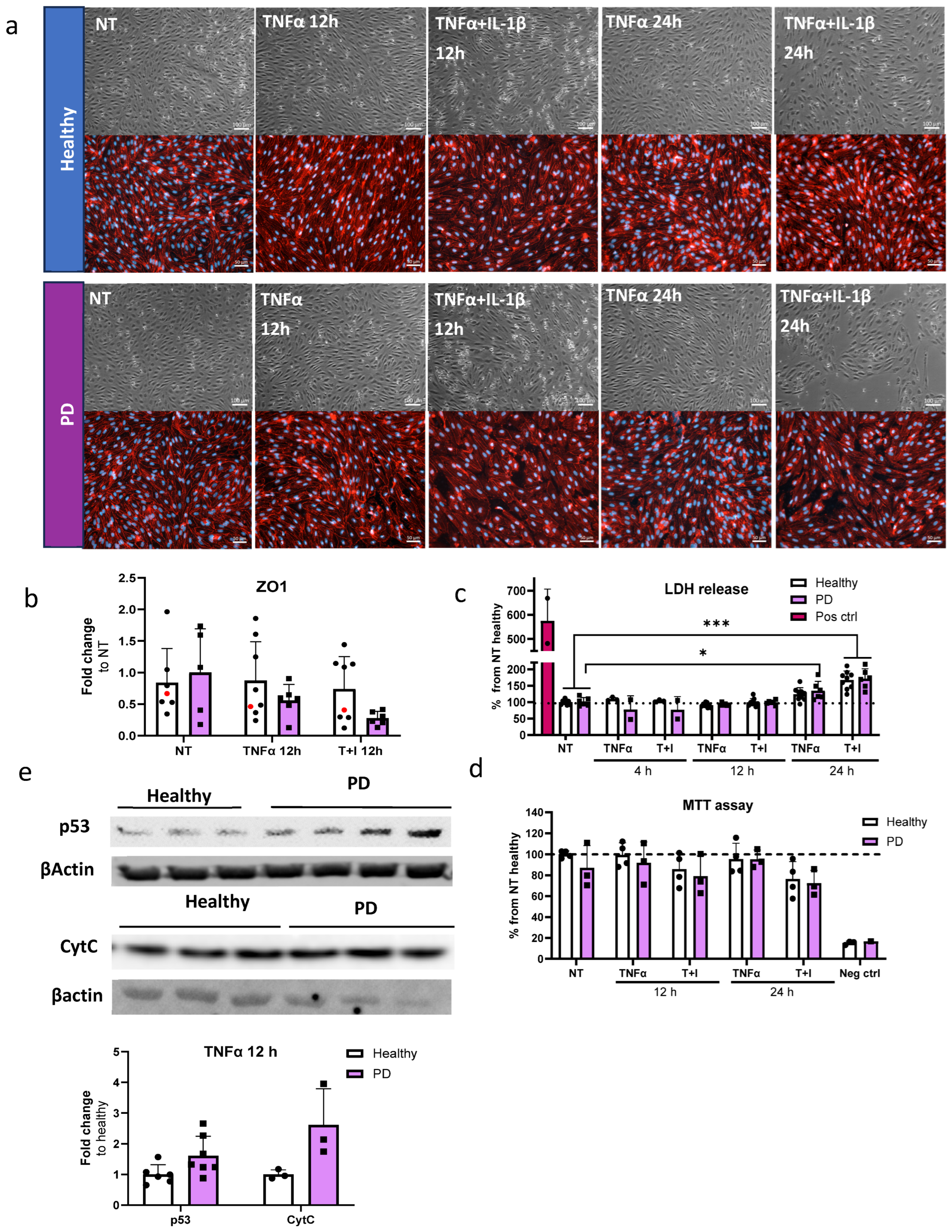
2.6. Prolonged Inflammatory Environment Affects Endothelial Cells Viability
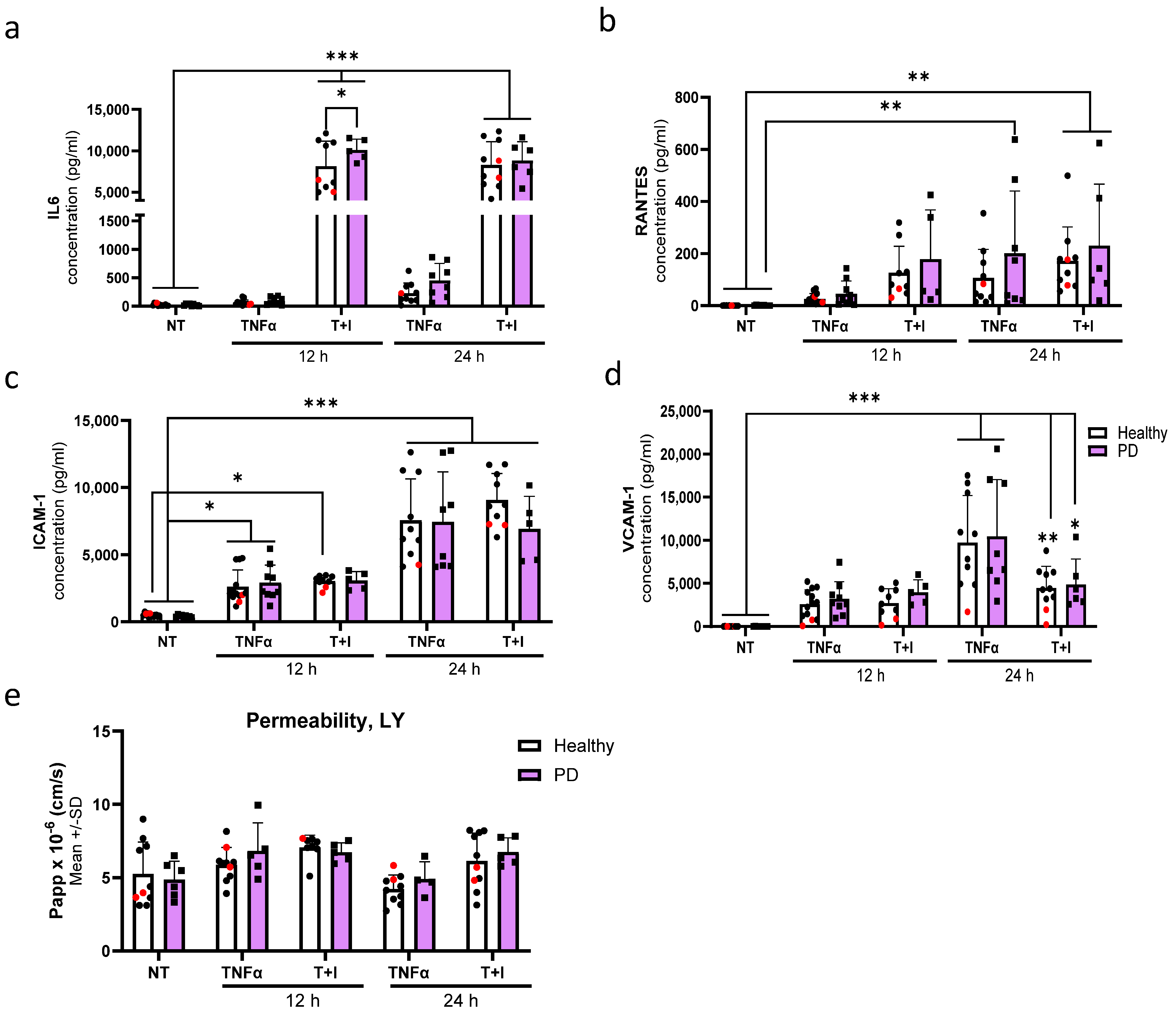
2.7. Endothelial Cells Show Aberrant Phenotype in Pro-Inflammatory Conditions
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Cell Culture
4.2. Endothelial Cell Differentiation
4.3. Immunocytochemistry
4.4. RT-qPCR
4.5. α-Synuclein ELISA
4.6. Seahorse
4.7. Inflmammatory Exposures
4.8. Total RNA Sequencing
4.9. LDH Cytotoxicity Test
4.10. MTT Assay
4.11. Western Blotting
4.12. Quantification of Immunofluorescence
4.13. Cytometric Bead Assay
4.14. Permeability Assay
4.15. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinson’s Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Piramanayagam, S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019, 6, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current Understanding of the Molecular Mechanisms in Parkinson’s Disease: Targets for Potential Treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.M.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.H.; Portman, A.; Leenders, K.L. Decreased blood–brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; Van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Kim, H.; Shin, J.-Y.; Lee, Y.-S.; Yun, S.P.; Maeng, H.-J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int. J. Mol. Sci. 2020, 21, 8538. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.H.; Yi, H.; Sunwoo, M.K.; Hong, J.Y.; Sohn, Y.H.; Lee, P.H. Cerebral microbleeds in patients with Parkinson’s disease. J. Neurol. 2014, 261, 1628–1635. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.; Parkes, L. PO071 Mri assessment of neurovascular changes in idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, A30. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal Blood–Brain Barrier Permeability in Parkinson’s Disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood–brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; Bjerkén, S.A.; Marcellino, D.; Virel, A. An altered blood–brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson’s disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Kim, Y.-S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 Contributes to Nigrostriatal Dopaminergic Neuronal Loss, BBB Damage, and Neuroinflammation in an MPTP Mouse Model of Parkinson’s Disease. Mediat. Inflamm. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-α knockout and minocycline treatment attenuates blood–brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef]
- Hongge, L.; Kexin, G.; Xiaojie, M.; Nian, X.; Jinsha, H. The Role of LRRK2 in the Regulation of Monocyte Adhesion to Endothelial Cells. J. Mol. Neurosci. 2014, 55, 233–239. [Google Scholar] [CrossRef]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood–brain barrier. Exp. Neurol. 2004, 190, 446–455. [Google Scholar] [CrossRef]
- Harding, A.; Cortez-Toledo, E.; Magner, N.L.; Beegle, J.R.; Coleal-Bergum, D.P.; Hao, D.; Wang, A.; Nolta, J.A.; Zhou, P. Highly Efficient Differentiation of Endothelial Cells from Pluripotent Stem Cells Requires the MAPK and the PI3K Pathways. Stem Cells 2017, 35, 909–919. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Ohtonen, S.; Giudice, L.; Jäntti, H.; Fazaludeen, M.F.; Shakirzyanova, A.; Gómez-Budia, M.; Välimäki, N.-N.; Niskanen, J.; Korvenlaita, N.; Fagerlund, I.; et al. Human iPSC-derived microglia carrying the LRRK2-G2019S mutation show a Parkinson’s disease related transcriptional profile and function. Sci. Rep. 2023, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, L.; Qin, L.; Huang, H.; Li, X. Single-Cell Transcriptomics Uncovers Cellular Heterogeneity, Mechanisms, and Therapeutic Targets for Parkinson’s Disease. Front. Genet. 2022, 13, 686739. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.Y.; Golmohammadi, M.; Amin, R.S.; Bustani, G.S.; Romero-Parra, R.M.; Zabibah, R.S.; Oz, T.; Jalil, A.T.; Soltani, A.; Kujawska, M. Therapeutic Targeting of Krüppel-Like Factor 4 and Its Pharmacological Potential in Parkinson’s Disease: A Comprehensive Review. Mol. Neurobiol. 2023, 61, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Tugal, D.; Jain, M.K.; Simon, D.I. Endothelial KLF4: Crippling Vascular Injury? J. Am. Hear. Assoc. 2014, 3, e000769. [Google Scholar] [CrossRef]
- Huang, T.; Yin, J.; Ren, S.; Zhang, X. Protective effects of KLF4 on blood–brain barrier and oxidative stress after cerebral ischemia–reperfusion in rats through the Nrf2/Trx1 pathway. Cytokine 2023, 169, 156288. [Google Scholar] [CrossRef]
- Yang, H.; Xi, X.; Zhao, B.; Su, Z.; Wang, Z. KLF4 protects brain microvascular endothelial cells from ischemic stroke induced apoptosis by transcriptionally activating MALAT1. Biochem. Biophys. Res. Commun. 2018, 495, 2376–2382. [Google Scholar] [CrossRef]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerszten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like Factor 4 Regulates Endothelial Inflammation. J. Biol. Chem. 2007, 282, 13769–13779. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zheng, S.; Lu, M. Downregulation of lncRNA MEG3 is involved in Parkinson’s disease. Metab. Brain Dis. 2021, 36, 2323–2328. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, J.; Wang, S.; Zhao, J. Association of the Plasma Long Non-coding RNA MEG3 With Parkinson’s Disease. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tamizkar, K.H.; Gorji, P.; Gholipour, M.; Hussen, B.M.; Mazdeh, M.; Eslami, S.; Taheri, M.; Ghafouri-Fard, S. Parkinson’s Disease Is Associated with Dysregulation of Circulatory Levels of lncRNAs. Front. Immunol. 2021, 12, 763323. [Google Scholar] [CrossRef]
- Peltonen, S.; Sonninen, T.-M.; Niskanen, J.; Koistinaho, J.; Ruponen, M.; Lehtonen, Š. Mutated LRRK2 induces a reactive phenotype and alters migration in human iPSC-derived pericyte-like cells. Fluids Barriers CNS 2024, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Li, Y.-J.; Li, D.-J.; Li, P.; Wang, J.-Y.; Diao, Y.-P.; Ye, G.-D.; Li, Y.-F. Long noncoding RNA MEG3 prevents vascular endothelial cell senescence by impairing miR-128-dependent Girdin downregulation. Am. J. Physiol. Physiol. 2019, 316, C830–C843. [Google Scholar] [CrossRef]
- Ruan, W.; Zhao, F.; Zhao, S.; Zhang, L.; Shi, L.; Pang, T. Knockdown of long noncoding RNA MEG3 impairs VEGF-stimulated endothelial sprouting angiogenesis via modulating VEGFR2 expression in human umbilical vein endothelial cells. Gene 2018, 649, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.-Z.; Tian, W.; Fu, H.-T.; Li, C.-P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Song, J.; Huang, S.; Wang, K.; Li, W.; Pao, L.; Chen, F.; Zhao, X. Long Non-coding RNA MEG3 Attenuates the Angiotensin II-Induced Injury of Human Umbilical Vein Endothelial Cells by Interacting With p53. Front. Genet. 2019, 10, 78. [Google Scholar] [CrossRef]
- Cheng, X.; Ali, M.S.S.H.; Moran, M.; Viana, M.P.; Schlichte, S.L.; Zimmerman, M.C.; Khalimonchuk, O.; Feinberg, M.W.; Sun, X. Long non-coding RNA Meg3 deficiency impairs glucose homeostasis and insulin signaling by inducing cellular senescence of hepatic endothelium in obesity. Redox Biol. 2021, 40, 101863. [Google Scholar] [CrossRef]
- Yang, P.; Min, X.-L.; Mohammadi, M.; Turner, C.; Faull, R.; Waldvogel, H.; Dragunow, M.; Guan, J. Endothelial Degeneration of Parkinson’s Disease Is Related to Alpha-Synuclein Aggregation. J. Alzheimers Dis. Parkinsonism 2017, 7, 370. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Zhao, Y.; Keshiya, S.; Perera, G.; Schramko, L.; Halliday, G.M.; Dzamko, N. LRRK2 kinase inhibitors reduce alpha-synuclein in human neuronal cell lines with the G2019S mutation. Neurobiol. Dis. 2020, 144, 105049. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Fukunaga, K. Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 17037. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to Long Chain Polyunsaturated Fatty Acids Triggers Rapid Multimerization of Synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. α-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of α-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary fat intake and risk of Parkinson’s disease: A case-control study in Japan. J. Neurol. Sci. 2009, 288, 117–122. [Google Scholar] [CrossRef]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological Rescue of Mitochondrial Deficits in iPSC-Derived Neural Cells from Patients with Familial Parkinson’s Disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Diaz, K.; Kohut, M.; Russell, D.; Stegemöller, E. Peripheral inflammatory cytokines and motor symptoms in persons with Parkinson’s disease. Brain Behav. Immun. Health 2022, 21, 100442. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.W.; Schuh, A.F.S.; Saute, J.; Townsend, R.; Fricke, D.; Leke, R.; Souza, D.O.; Portela, L.V.; Chaves, M.L.F.; Rieder, C.R.M. Interleukin-6 Serum Levels in Patients with Parkinson’s Disease. Neurochem. Res. 2009, 34, 1401–1404. [Google Scholar] [CrossRef]
- Dufek, M.; Rektorova, I.; Thon, V.; Lokaj, J.; Rektor, I. Interleukin-6 May Contribute to Mortality in Parkinson’s Disease Patients: A 4-Year Prospective Study. Parkinson’s Dis. 2015, 2015, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Serum levels of interleukin-6 are elevated in patients with Parkinson’s disease and correlate with physical performance. Neurosci. Lett. 2010, 468, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Li, H.; Pan, X.; Wang, Y. New Insights into the Roles of p53 in Central Nervous System Diseases. Int. J. Neuropsychopharmacol. 2023, 26, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-W.; Li, G.-R.; Zhang, B.-L.; Feng, J.-J.; Zhao, H. Damage to dopaminergic neurons is mediated by proliferating cell nuclear antigen through the p53 pathway under conditions of oxidative stress in a cell model of Parkinson’s disease. Int. J. Mol. Med. 2015, 37, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neurosci. Lett. 2006, 414, 94–97. [Google Scholar] [CrossRef]
- Sekar, S.; Taghibiglou, C. Nuclear accumulation of GAPDH, GluA2 and p53 in post-mortem substantia nigral region of patients with Parkinson’s disease. Neurosci. Lett. 2020, 716, 134641. [Google Scholar] [CrossRef]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 Induces Apoptosis by Caspase Activation through Mitochondrial Cytochrome c Release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef]
- Yan, H.; Yuan, J.; Gao, L.; Rao, J.; Hu, J. Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience 2016, 337, 191–199. [Google Scholar] [CrossRef]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast–Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Shihabudeen Haider Ali, M.S.; Cheng, X.; Moran, M.; Haemmig, S.; Naldrett, M.J.; Alvarez, S.; Feinberg, M.W.; Sun, X. LncRNA Meg3 protects endothelial function by regulating the DNA damage response. Nucleic Acids Res. 2018, 47, 1505–1522. [Google Scholar] [CrossRef]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Gastfriend, B.D.; Soldati, S.; Perriot, S.; Mathias, A.; Sano, Y.; Shimizu, F.; Gosselet, F.; Kanda, T.; Palecek, S.P.; et al. Advancing human induced pluripotent stem cell-derived blood-brain barrier models for studying immune cell interactions. FASEB J. 2020, 34, 16693–16715. [Google Scholar] [CrossRef] [PubMed]
- Delsing, L.; Dönnes, P.; Sánchez, J.; Clausen, M.; Voulgaris, D.; Falk, A.; Herland, A.; Brolén, G.; Zetterberg, H.; Hicks, R.; et al. Barrier Properties and Transcriptome Expression in Human iPSC-Derived Models of the Blood–Brain Barrier. Stem Cells 2018, 36, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.M.; Houghton, S.; Magdeldin, T.; Durán, J.G.B.; Minotti, A.P.; Snead, A.; Sproul, A.; Nguyen, D.-H.T.; Xiang, J.; Fine, H.A.; et al. Pluripotent stem cell-derived epithelium misidentified as brain microvascular endothelium requires ETS factors to acquire vascular fate. Proc. Natl. Acad. Sci. USA 2021, 118, e2016950118. [Google Scholar] [CrossRef]
- Ozgür, B.; Puris, E.; Brachner, A.; Appelt-Menzel, A.; Oerter, S.; Balzer, V.; Holst, M.R.; Christiansen, R.F.; Hyldig, K.; Buckley, S.T.; et al. Characterization of an iPSC-based barrier model for blood-brain barrier investigations using the SBAD0201 stem cell line. Fluids Barriers CNS 2023, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Lehtonen, Š.; Chumarina, M.; A Puttonen, K.; Azevedo, C.; Lebedeva, O.; Ruponen, M.; Oksanen, M.; Djelloul, M.; Collin, A.; et al. Creation of a library of induced pluripotent stem cells from Parkinsonian patients. npj Parkinsons Dis. 2016, 2, 16009. [Google Scholar] [CrossRef]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonninen, T.-M.; Peltonen, S.; Niskanen, J.; Hämäläinen, R.H.; Koistinaho, J.; Lehtonen, Š. LRRK2 G2019S Mutated iPSC-Derived Endothelial Cells Exhibit Increased α-Synuclein, Mitochondrial Impairment, and Altered Inflammatory Responses. Int. J. Mol. Sci. 2024, 25, 12874. https://doi.org/10.3390/ijms252312874
Sonninen T-M, Peltonen S, Niskanen J, Hämäläinen RH, Koistinaho J, Lehtonen Š. LRRK2 G2019S Mutated iPSC-Derived Endothelial Cells Exhibit Increased α-Synuclein, Mitochondrial Impairment, and Altered Inflammatory Responses. International Journal of Molecular Sciences. 2024; 25(23):12874. https://doi.org/10.3390/ijms252312874
Chicago/Turabian StyleSonninen, Tuuli-Maria, Sanni Peltonen, Jonna Niskanen, Riikka H. Hämäläinen, Jari Koistinaho, and Šárka Lehtonen. 2024. "LRRK2 G2019S Mutated iPSC-Derived Endothelial Cells Exhibit Increased α-Synuclein, Mitochondrial Impairment, and Altered Inflammatory Responses" International Journal of Molecular Sciences 25, no. 23: 12874. https://doi.org/10.3390/ijms252312874
APA StyleSonninen, T.-M., Peltonen, S., Niskanen, J., Hämäläinen, R. H., Koistinaho, J., & Lehtonen, Š. (2024). LRRK2 G2019S Mutated iPSC-Derived Endothelial Cells Exhibit Increased α-Synuclein, Mitochondrial Impairment, and Altered Inflammatory Responses. International Journal of Molecular Sciences, 25(23), 12874. https://doi.org/10.3390/ijms252312874