
Morphometric Similarity Patterning of Amyloid-β and Tau Proteins Correlates with Transcriptomics in the Alzheimer’s Disease Continuum
 , , , , , , and
for the Alzheimer’s Disease Neuroimaging Initiative
, , , , , , and
for the Alzheimer’s Disease Neuroimaging Initiative
Abstract
1. Introduction
2. Results
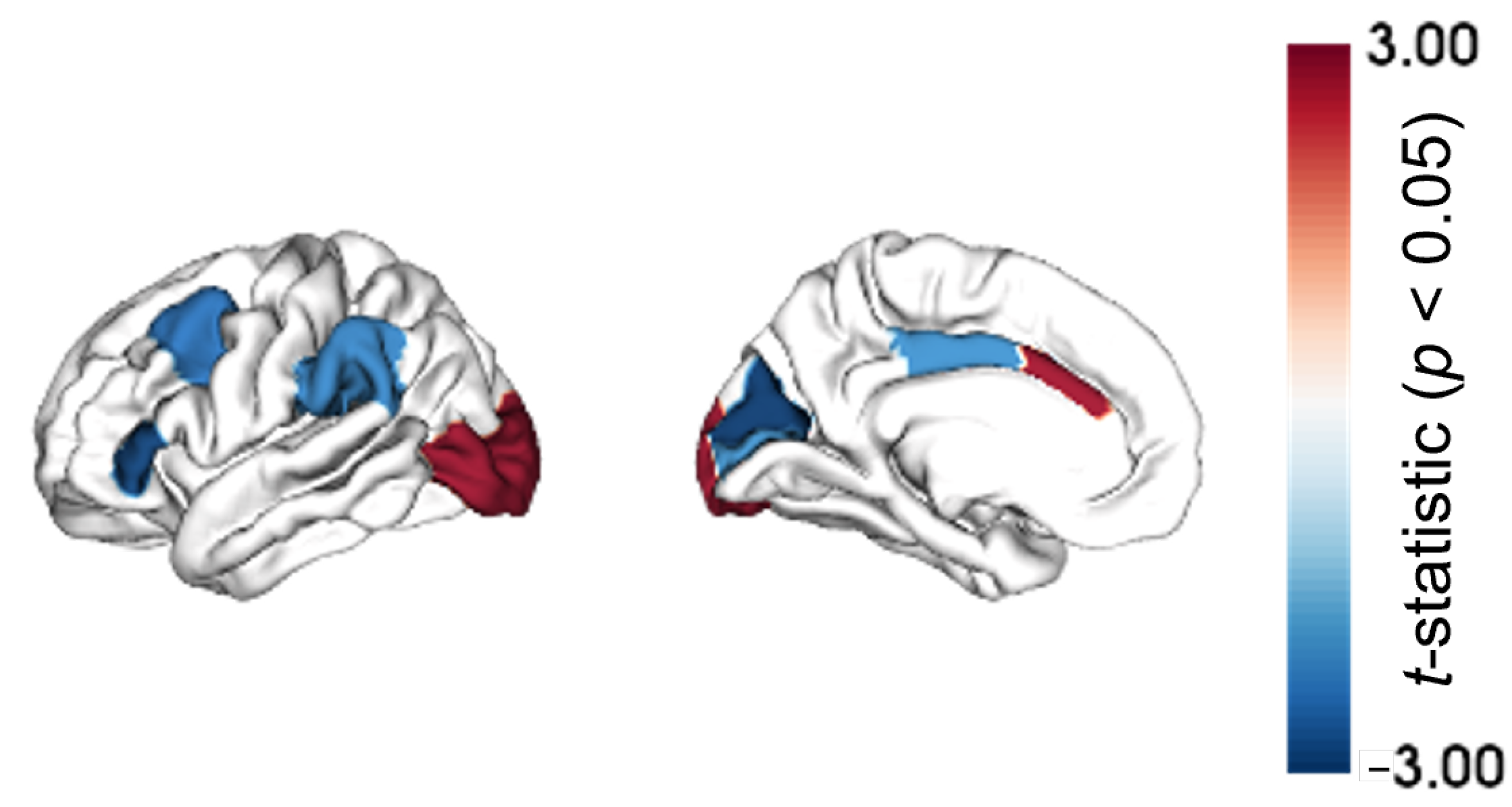
2.1. Regional Morphometric Similarity Group-Wise Differences
2.2. Regional Morphometric Similarity and Transcriptomics Relationship
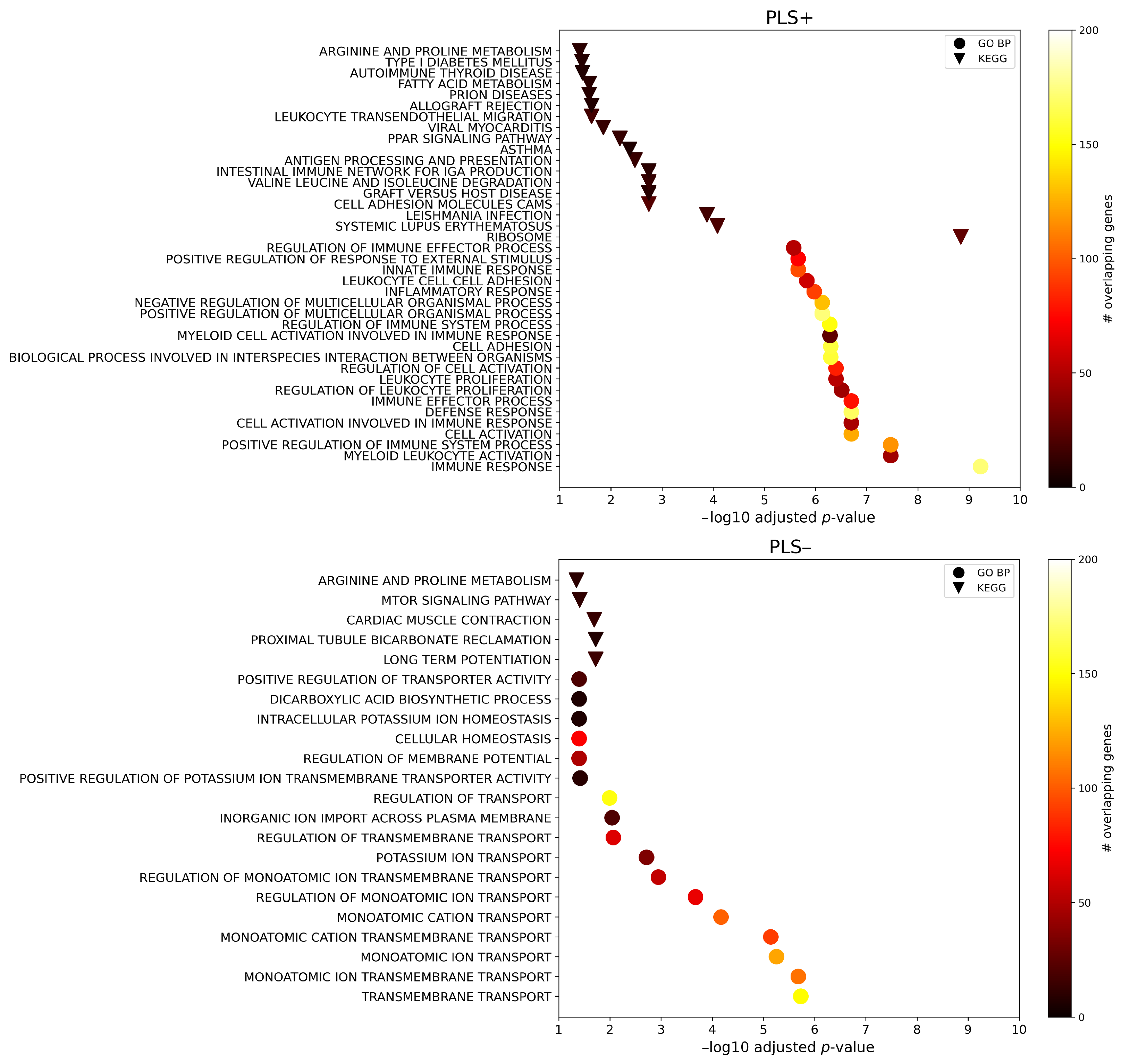
2.3. Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Data Cohort
4.2. Magnetic Resonance Imaging Data Processing
4.3. Regional Morphometric Similarity Group-Wise Differences
4.4. Gene Expression: Allen Human Brain Atlas
4.5. Regional Morphometric Similarity and Transcriptomics Relationship
4.6. Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MRI | Magnetic resonance imaging |
| PET | Positron emission tomography |
| A | Amyloid- |
| AD | Alzheimer’s disease |
| T1w | T1-weighted |
| dMRI | Diffusion MRI |
| AHBA | Allen Human Brain Atlas |
| MS | Morphometric similarity |
| CN | Cognitively unimpaired |
| MCI | Mildly cognitively impaired |
| MSN | MS network |
| SMC | Significant memory concern |
| A+/tau+ | A-positive/tau-positive |
| A−/tau− | A-negative/tau-negative |
| ADNI | AD Neuroimaging Initiative |
| MMSE | Mini-mental state examination |
| ROI | Region of interest |
| FDR | False discovery rate |
| PLS | Partial least squares |
| FUMA GWAS | Functional Mapping and Annotation of Genome-Wide Association Studies |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO BP | Gene Ontology biological process |
| PPI | Protein–protein interaction |
| OR | Odds ratio |
References
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Vilkaite, G.; Vogel, J.; Mattsson-Carlgren, N. Integrating amyloid and tau imaging with proteomics and genomics in Alzheimer’s disease. Cell Rep. Med. 2024, 5, 101735. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Risacher, S.L.; Nho, K.T.; Wen, Q.; Oblak, A.L.; Unverzagt, F.W.; Apostolova, L.G.; Farlow, M.R.; Brosch, J.R.; Clark, D.G.; et al. Spatial transcriptomic patterns underlying amyloid-β and tau pathology are associated with cognitive dysfunction in Alzheimer’s disease. Cell Reports 2024, 43, 113691. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Sepulcre, J.; Gonzalez-Escamilla, G.; Jelistratova, I.; Schöll, M.; Hansson, O.; Teipel, S.J. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 2018, 141, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Palmqvist, S.; Stomrud, E.; Vogel, J.; Hansson, O. Staging β-Amyloid Pathology with Amyloid Positron Emission Tomography. JAMA Neurol. 2019, 76, 1319–1329. [Google Scholar] [CrossRef]
- Sanchez-Rodriguez, L.M.; Khan, A.F.; Adewale, Q.; Bezgin, G.; Therriault, J.; Fernandez-Arias, J.; Servaes, S.; Rahmouni, N.; Tissot, C.; Stevenson, J.; et al. In-vivo neuronal dysfunction by Aβ and tau overlaps with brain-wide inflammatory mechanisms in Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1383163. [Google Scholar] [CrossRef]
- Pini, L.; Pievani, M.; Bocchetta, M.; Altomare, D.; Bosco, P.; Cavedo, E.; Galluzzi, S.; Marizzoni, M.; Frisoni, G.B. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res. Rev. 2016, 30, 25–48. [Google Scholar] [CrossRef]
- Le Bihan, D.; Mangin, J.-F.; Poupon, C.; Clark, C.A.; Pappata, S.; Molko, N.; Chabriat, H. Diffusion tensor imaging: Concepts and applications. J. Magn. Reson. Imaging 2001, 13, 534–546. [Google Scholar] [CrossRef]
- Spotorno, N.; Strandberg, O.; Vis, G.; Stomrud, E.; Nilsson, M.; Hansson, O. Measures of cortical microstructure are linked to amyloid pathology in Alzheimer’s disease. Brain 2022, 146, 1602–1614. [Google Scholar] [CrossRef]
- Brusini, L.; Cruciani, F.; Dall’Aglio, G.; Zajac, T.; Boscolo Galazzo, I.; Zucchelli, M.; Menegaz, G. XAI-Based Assessment of the AMURA Model for Detecting Amyloid-β and Tau Microstructural Signatures in Alzheimer’s Disease. IEEE J. Transl. Eng. Health Med. 2024, 12, 569–579. [Google Scholar] [CrossRef]
- Bassett, D.S.; Bullmore, E.; Verchinski, B.A.; Mattay, V.S.; Weinberger, D.R.; Meyer-Lindenberg, A. Hierarchical Organization of Human Cortical Networks in Health and Schizophrenia. J. Neurosci. 2008, 28, 9239–9248. [Google Scholar] [CrossRef] [PubMed]
- Seidlitz, J.; Vasa, F.; Shinn, M.; Romero-Garcia, R.; Whitaker, K.J.; Vértes, P.E.; Wagstyl, K.; Kirkpatrick Reardon, P.; Clasen, L.; Liu, S.; et al. Morphometric Similarity Networks Detect Microscale Cortical Organization and Predict Inter-Individual Cognitive Variation. Neuron 2018, 97, 231–247.e7. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.E.; Seidlitz, J.; Whitaker, K.J.; Romero-Garcia, R.; Clifton, N.E.; Scarpazza, C.; van Amelsvoort, T.; Marcelis, M.; van Os, J.; Donohoe, G.; et al. Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc. Natl. Acad. Sci. USA 2019, 116, 9604–9609. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Keller, S.S.; Seidlitz, J.; Chen, H.; Li, B.; Weng, Y.; Meng, Y.; Yang, S.; Xu, Q.; Zhang, Q.; et al. Cortical morphometric vulnerability to generalised epilepsy reflects chromosome- and cell type-specific transcriptomic signatures. Neuropathol. Appl. Neurobiol. 2023, 49, e12857. [Google Scholar] [CrossRef]
- Lei, W.; Xiao, Q.; Wang, C.; Gao, W.; Xiao, Y.; Dai, Y.; Lu, G.; Su, L.; Zhong, Y. Cell-type-specific genes associated with cortical structural abnormalities in pediatric bipolar disorder. Psychoradiology 2022, 2, 56–65. [Google Scholar] [CrossRef]
- Martins, D.; Dipasquale, O.; Veronese, M.; Turkheimer, F.; Loggia, M.L.; McMahon, S.; Howard, M.A.; Williams, S.C.R. Transcriptional and cellular signatures of cortical morphometric remodelling in chronic pain. Pain 2022, 163, e759–e773. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, F.; Lei, W.; Ke, J.; Dai, Y.; Qi, R.; Lu, G.; Zhong, Y. Transcriptional signal and cell specificity of genes related to cortical structural differences of post-traumatic stress disorder. J. Psychiatr. Res. 2023, 160, 28–37. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Xing, Y.; Yu, M.; Wang, X.; Ren, J.; Liu, W.; Zhong, Y. Morphometric similarity differences in drug-naive Parkinson’s disease correlate with transcriptomic signatures. CNS Neurosci. Ther. 2024, 30, e14680. [Google Scholar] [CrossRef]
- Long, J.; Li, J.; Xie, B.; Jiao, Z.; Shen, G.; Liao, W.; Song, X.; Le, H.; Xia, J.; Wu, S. Morphometric similarity network alterations in COVID-19 survivors correlate with behavioral features and transcriptional signatures. Neuroimage Clin. 2023, 39, 103498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, M.; Xie, Z.; Wu, H.; Zhang, N.; Shen, J. Bridging the Gap Between Morphometric Similarity Mapping and Gene Transcription in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 731292. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Barthel, H.; Sepulcre, J.; Dyrba, M.; Sabri, O.; Teipel, S.J. In vivo staging of regional amyloid deposition. Neurology 2017, 89, 2031–2038. [Google Scholar] [CrossRef]
- Hwang, U.; Kim, S.W.; Jung, D.; Kim, S.; Lee, H.; Seo, S.W.; Seong, J.K.; Yoon, S. Real-world prediction of preclinical Alzheimer’s disease with a deep generative model. Artif. Intell. Med. 2023, 144, 102654. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Guillozet, A.L.; Mesulam, M.M.; Smiley, J.F.; Mash, D.C. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef]
- Darvesh, S.; Kumar, R.; Roberts, S.; Walsh, R.; Martin, E. Butyrylcholinesterase-Mediated Enhancement of the Enzymatic Activity of Trypsin. Cell. Mol. Neurobiol. 2001, 21, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Martins-de-Souza, D.; Guest, P.C.; Mann, D.M.; Roeber, S.; Rahmoune, H.; Bauder, C.; Kretzschmar, H.; Volk, B.; Baborie, A.; Bahn, S. Proteomic Analysis Identifies Dysfunction in Cellular Transport, Energy, and Protein Metabolism in Different Brain Regions of Atypical Frontotemporal Lobar Degeneration. J. Proteome Res. 2012, 11, 2533–2543. [Google Scholar] [CrossRef]
- Rajesh, Y.; Kanneganti, T.D. Innate Immune Cell Death in Neuroinflammation and Alzheimer’s Disease. Cells 2022, 11, 1885. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2018, 216, 60–70. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Alsema, A.M.; Jiang, Q.; Kracht, L.; Gerrits, E.; Dubbelaar, M.L.; Miedema, A.; Brouwer, N.; Hol, E.M.; Middeldorp, J.; van Dijk, R.; et al. Profiling Microglia From Alzheimer’s Disease Donors and Non-demented Elderly in Acute Human Postmortem Cortical Tissue. Front. Mol. Neurosci. 2020, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.T.; Taylor, D.; Kneynsberg, A.; Bodea, L.G.; Götz, J. Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol. Commun. 2021, 9, 110. [Google Scholar] [CrossRef]
- Zeng, H.; Huang, J.; Zhou, H.; Meilandt, W.J.; Dejanovic, B.; Zhou, Y.; Bohlen, C.J.; Lee, S.-H.; Ren, J.; Liu, A.; et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 430–446. [Google Scholar] [CrossRef]
- Cohen, S.; Cummings, J.; Knox, S.; Potashman, M.; Harrison, J. Clinical Trial Endpoints and Their Clinical Meaningfulness in Early Stages of Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 507–522. [Google Scholar] [CrossRef]
- Zavaliangos-Petropulu, A.; Nir, T.M.; Thomopoulos, S.I.; Reid, R.I.; Bernstein, M.A.; Borowski, B.; Jack, C.R., Jr.; Weiner, M.W.; Jahanshad, N.; Thompson, P.M. Diffusion MRI Indices and Their Relation to Cognitive Impairment in Brain Aging: The Updated Multi-protocol Approach in ADNI3. Front. Neuroinformatics 2019, 13, 2. [Google Scholar] [CrossRef]
- Thomas, K.R.; Bangen, K.J.; Rotblatt, L.J.; Weigand, A.J.; Edwards, L.; Tosun, D.; Galasko, D. Self- and study partner–reported cognitive decline in older adults without dementia: The role of α-synuclein and amyloid biomarkers in the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. 2024, 20, 7777–7787. [Google Scholar] [CrossRef]
- Jenkinson, M.; Beckmann, C.F.; Behrens, T.E.; Woolrich, M.W.; Smith, S.M. FSL. NeuroImage 2012, 62, 782–790. [Google Scholar] [CrossRef]
- Fischl, B.; Salat, D.H.; Busa, E.; Albert, M.; Dieterich, M.; Haselgrove, C.; van der Kouwe, A.; Killiany, R.; Kennedy, D.; Klaveness, S.; et al. Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron 1991, 33, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.L.; Sotiropoulos, S.N. An integrated approach to correction for off-resonance effects and subject movement in diffusion MR imaging. NeuroImage 2016, 125, 1063–1078. [Google Scholar] [CrossRef]
- Garyfallidis, E.; Brett, M.; Amirbekian, B.; Rokem, A.; Van Der Walt, S.; Descoteaux, M.; Nimmo-Smith, I. Dipy, a library for the analysis of diffusion MRI data. Front. Neuroinformatics 2014, 8, 8. [Google Scholar] [CrossRef]
- Veraart, J.; Fieremans, E.; Novikov, D.S. Diffusion MRI noise mapping using random matrix theory. Magn. Reson. Med. 2016, 76, 1582–1593. [Google Scholar] [CrossRef]
- Jenkinson, M.; Bannister, P.; Brady, M.; Smith, S. Improved Optimization for the Robust and Accurate Linear Registration and Motion Correction of Brain Images. NeuroImage 2002, 17, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Nir, T.M.; Jahanshad, N.; Villalon-Reina, J.E.; Isaev, D.; Zavaliangos-Petropulu, A.; Zhan, L.; Leow, A.D.; Jack, C.R., Jr.; Weiner, M.W.; Thompson, P.M. Fractional anisotropy derived from the diffusion tensor distribution function boosts power to detect Alzheimer’s disease deficits. Magn. Reson. Med. 2017, 78, 2322–2333. [Google Scholar] [CrossRef]
- Avants, B.B.; Tustison, N.J.; Stauffer, M.; Song, G.; Wu, B.; Gee, J.C. The Insight ToolKit image registration framework. Front. Neuroinformatics 2014, 8, 44. [Google Scholar] [CrossRef]
- Basser, P.J.; Pierpaoli, C. Microstructural and Physiological Features of Tissues Elucidated by Quantitative-Diffusion-Tensor MRI. J. Magn. Reson. Ser. B 1996, 111, 209–219. [Google Scholar] [CrossRef]
- Markello, R.D.; Arnatkeviciute, A.; Poline, J.-B.; Fulcher, B.D.; Fornito, A.; Misic, B. Standardizing workflows in imaging transcriptomics with the abagen toolbox. eLife 2021, 10, e72129. [Google Scholar] [CrossRef]
- Arnatkeviciute, A.; Fulcher, B.D.; Fornito, A. A practical guide to linking brain-wide gene expression and neuroimaging data. NeuroImage 2019, 189, 353–367. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.R.; Lobaugh, N.J. Partial least squares analysis of neuroimaging data: Applications and advances. NeuroImage 2004, 23, S250–S263. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Seidlitz, J.; Nadig, A.; Liu, S.; Bethlehem, R.A.I.; Vertes, P.E.; Morgan, S.E.; Vasa, F.; Romero-Garcia, R.; Lalonde, F.M.; Clasen, L.S.; et al. Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat. Commun. 2020, 11, 3358. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Santpere, G.; Imamura Kawasawa, Y.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Shen, L. GeneOverlap: Test and Visualize Gene Overlaps; R Package Version 1.40.0. 2024. Available online: https://bioconductor.org/packages/release/bioc/html/GeneOverlap.html (accessed on 25 November 2024).
- Himmelstein, D.S.; Baranzini, S.E. Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease-Associated Genes. PLoS Comput. Biol. 2015, 11, e1004259. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Ramirez-Anguita, J.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Covariate | A−/tau− | A+/tau+ | p-Value |
|---|---|---|---|
| Age [Y] | 71.05 (7.10) | 77.18 (7.92) | |
| Gender [M/F] | 71/101 | 60/66 | 0.148 |
| Education [Y] | 16.70 (2.38) | 16.01 (2.54) | 0.016 |
| MMSE | 28.85 (1.47) | 26.06 (4.62) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brusini, L.; Dolci, G.; Pini, L.; Cruciani, F.; Pizzagalli, F.; Provero, P.; Menegaz, G.; Boscolo Galazzo, I., for the Alzheimer’s Disease Neuroimaging Initiative. Morphometric Similarity Patterning of Amyloid-β and Tau Proteins Correlates with Transcriptomics in the Alzheimer’s Disease Continuum. Int. J. Mol. Sci. 2024, 25, 12871. https://doi.org/10.3390/ijms252312871
Brusini L, Dolci G, Pini L, Cruciani F, Pizzagalli F, Provero P, Menegaz G, Boscolo Galazzo I for the Alzheimer’s Disease Neuroimaging Initiative. Morphometric Similarity Patterning of Amyloid-β and Tau Proteins Correlates with Transcriptomics in the Alzheimer’s Disease Continuum. International Journal of Molecular Sciences. 2024; 25(23):12871. https://doi.org/10.3390/ijms252312871
Chicago/Turabian StyleBrusini, Lorenza, Giorgio Dolci, Lorenzo Pini, Federica Cruciani, Fabrizio Pizzagalli, Paolo Provero, Gloria Menegaz, and Ilaria Boscolo Galazzo for the Alzheimer’s Disease Neuroimaging Initiative. 2024. "Morphometric Similarity Patterning of Amyloid-β and Tau Proteins Correlates with Transcriptomics in the Alzheimer’s Disease Continuum" International Journal of Molecular Sciences 25, no. 23: 12871. https://doi.org/10.3390/ijms252312871
APA StyleBrusini, L., Dolci, G., Pini, L., Cruciani, F., Pizzagalli, F., Provero, P., Menegaz, G., & Boscolo Galazzo, I., for the Alzheimer’s Disease Neuroimaging Initiative. (2024). Morphometric Similarity Patterning of Amyloid-β and Tau Proteins Correlates with Transcriptomics in the Alzheimer’s Disease Continuum. International Journal of Molecular Sciences, 25(23), 12871. https://doi.org/10.3390/ijms252312871