
Oxidative Stress-Associated Alteration of circRNA and Their ceRNA Network in Differentiating Neuroblasts



Abstract
1. Introduction
2. Results
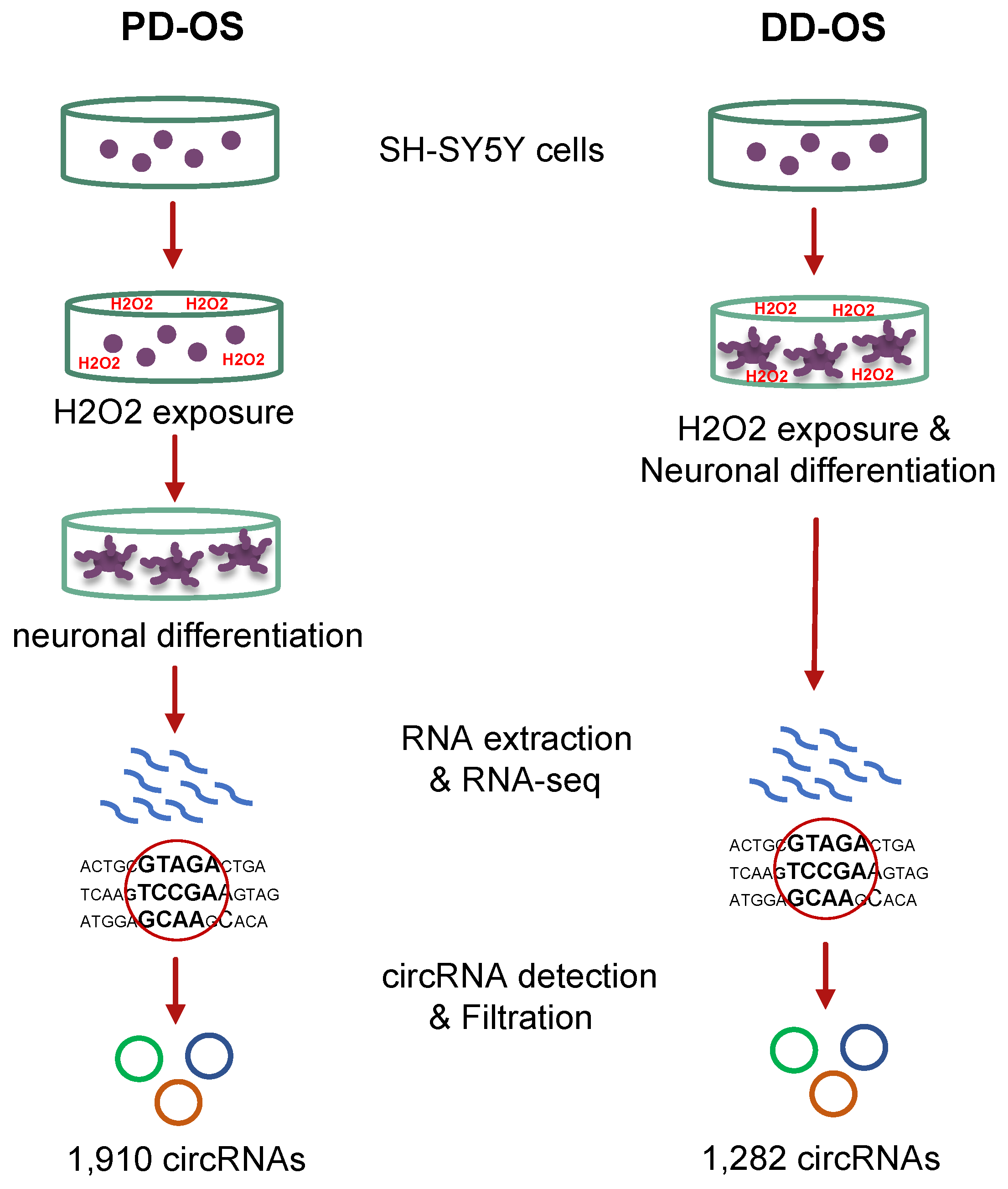
2.1. Oxidative Stress Models and Profiling the circRNAome
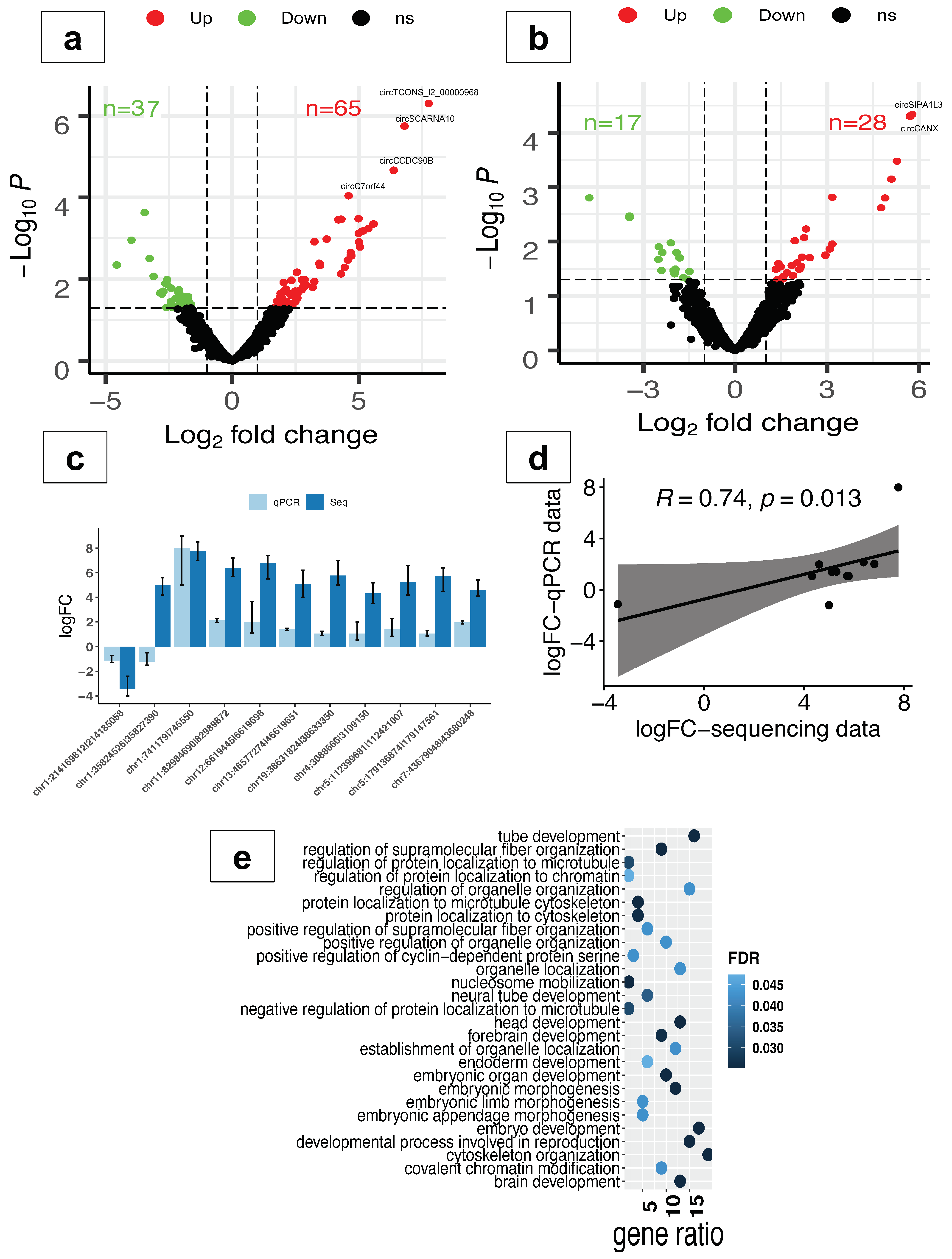
2.2. CircRNAs Are Differentially Expressed in Response to Oxidative Stress
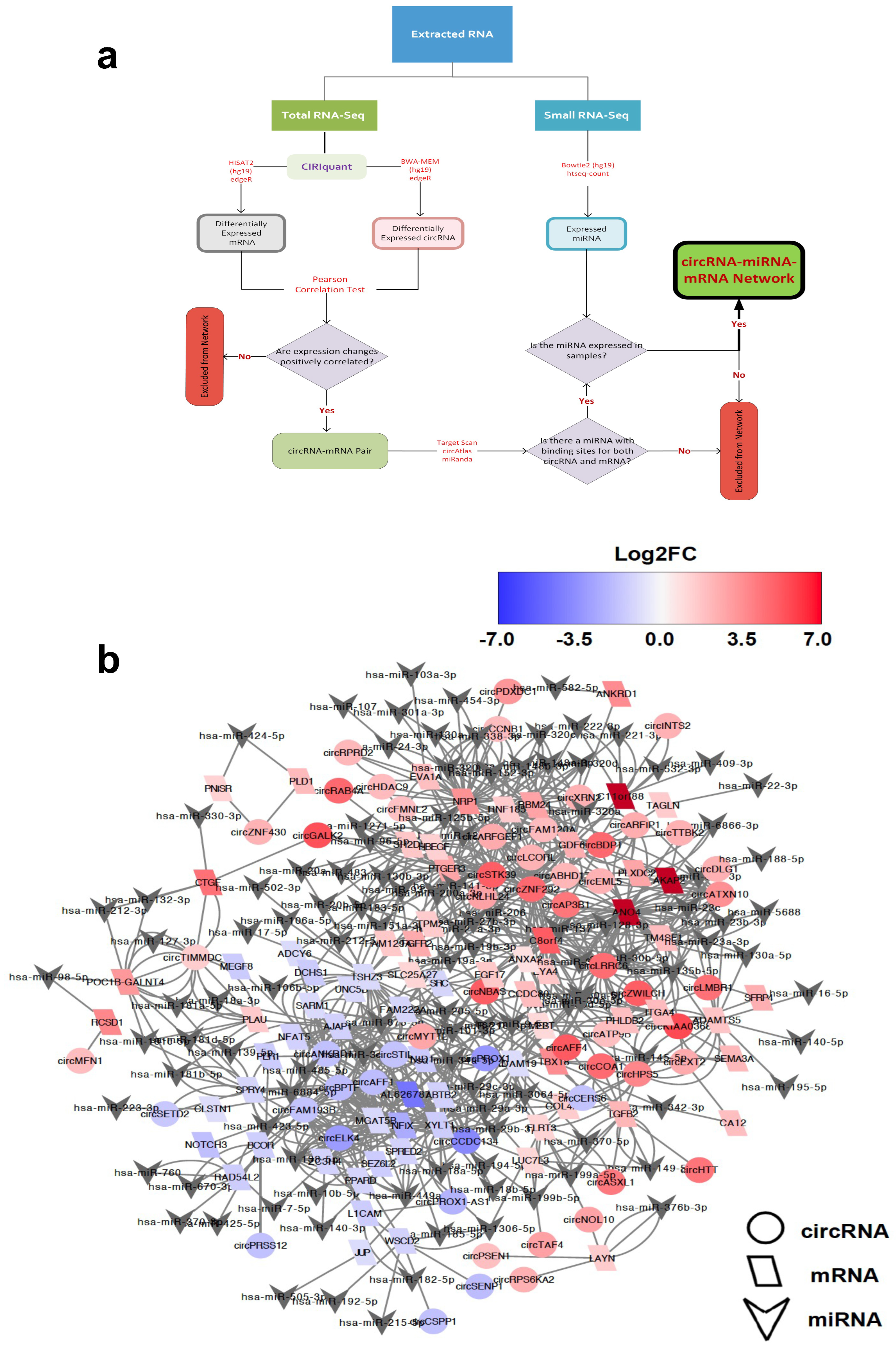
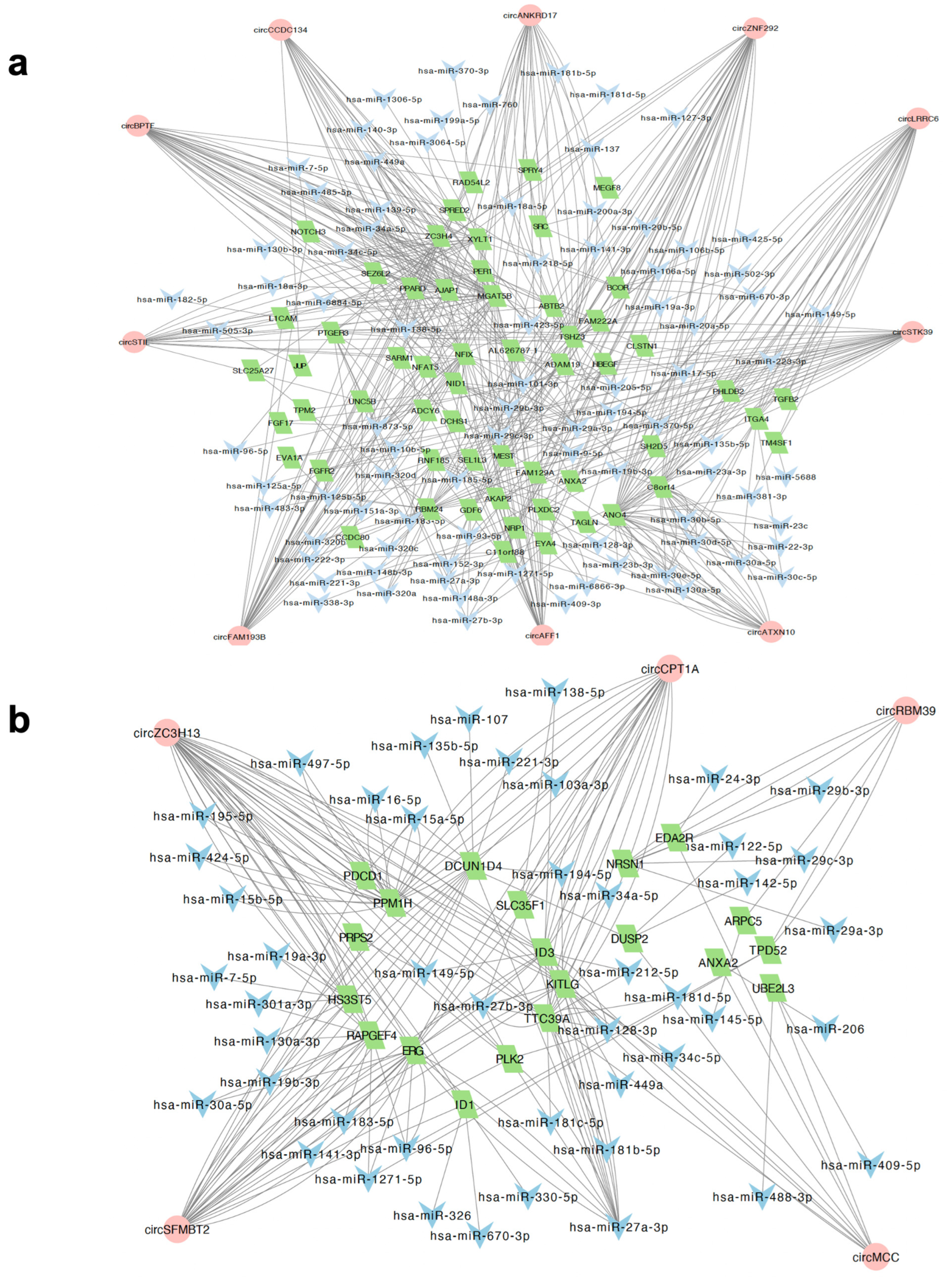
2.3. Identification of circRNA-Mediated ceRNA Network
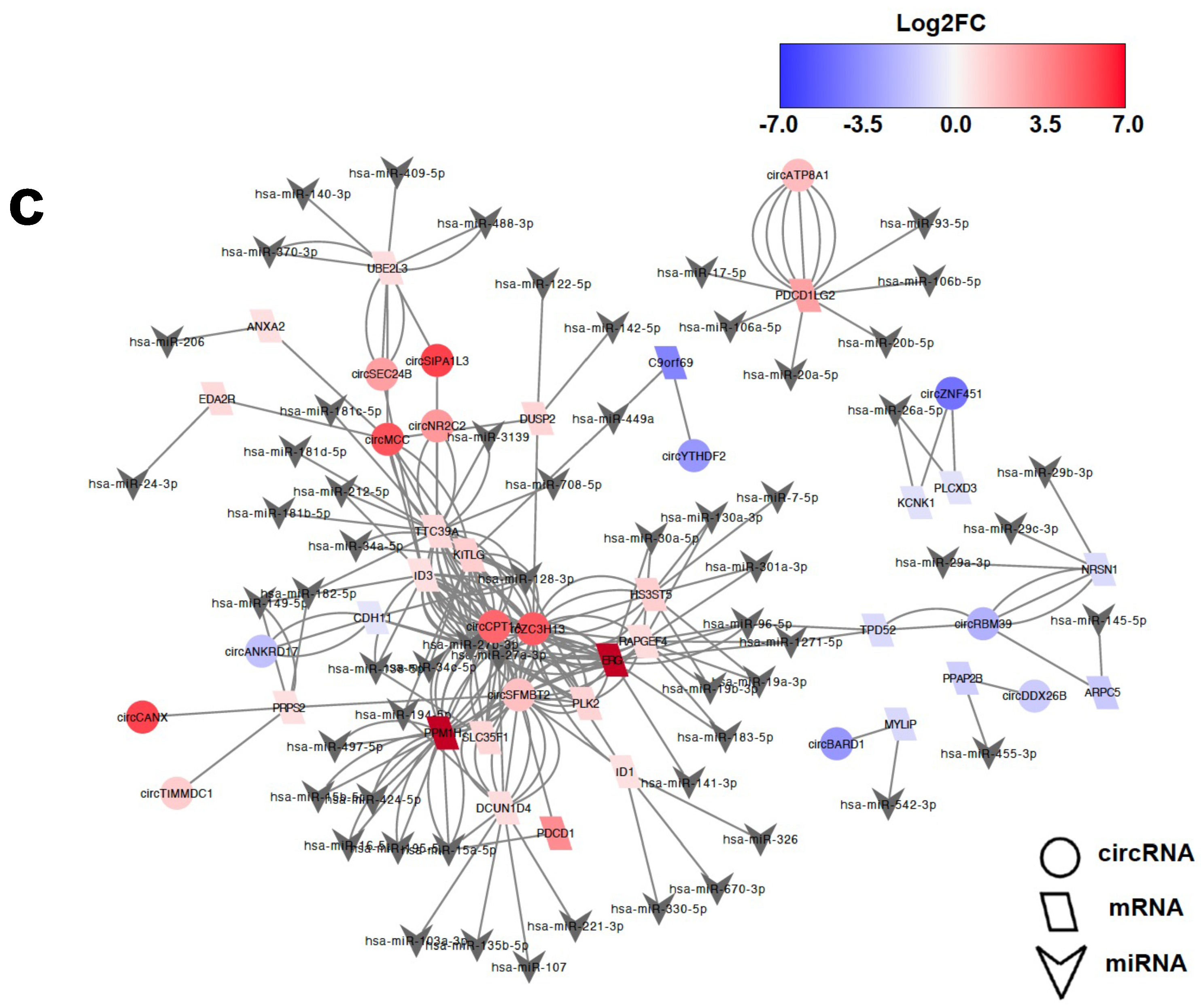
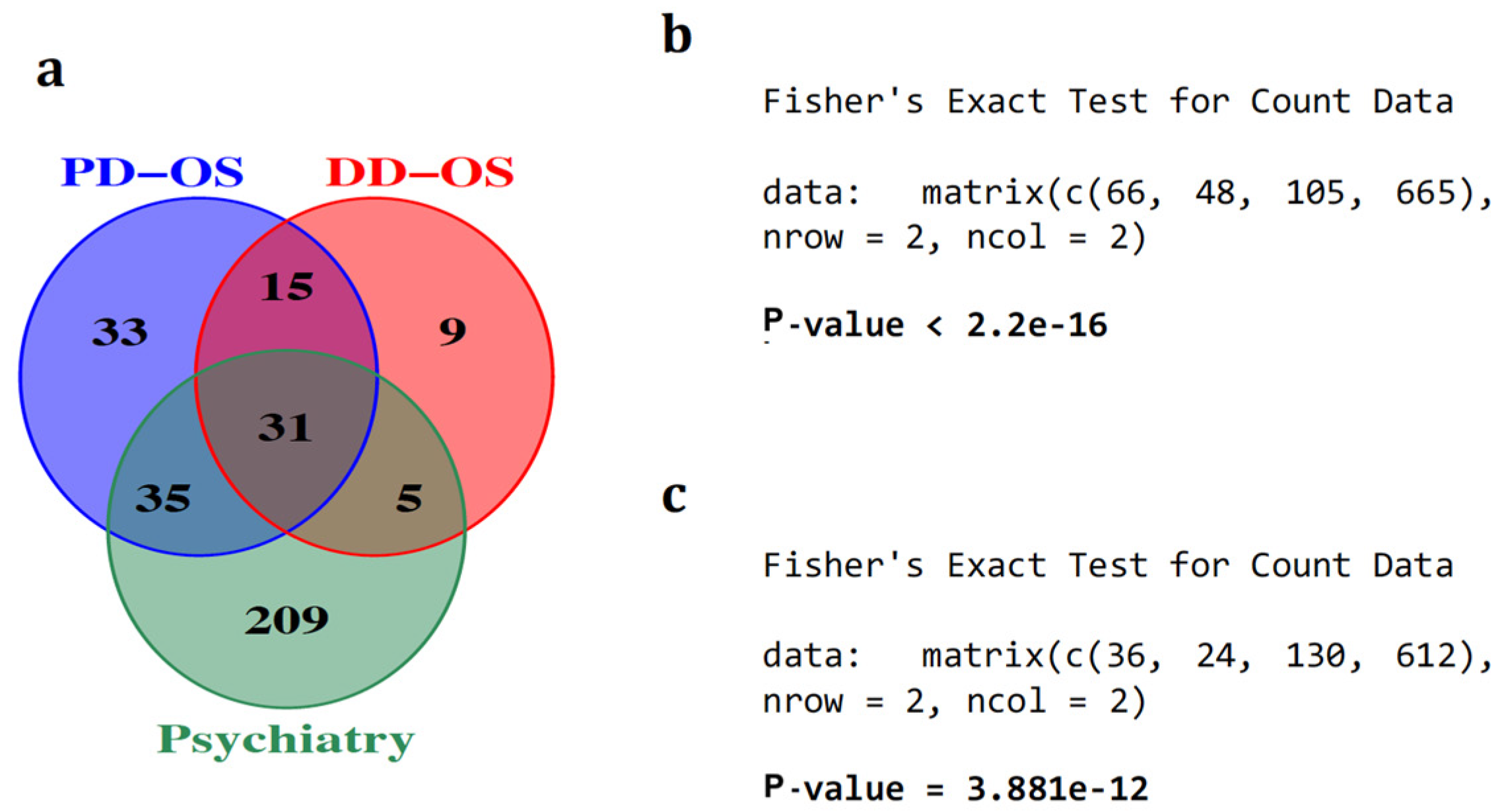
2.4. Characterisation of Hub circRNAs in the ceRNA Networks
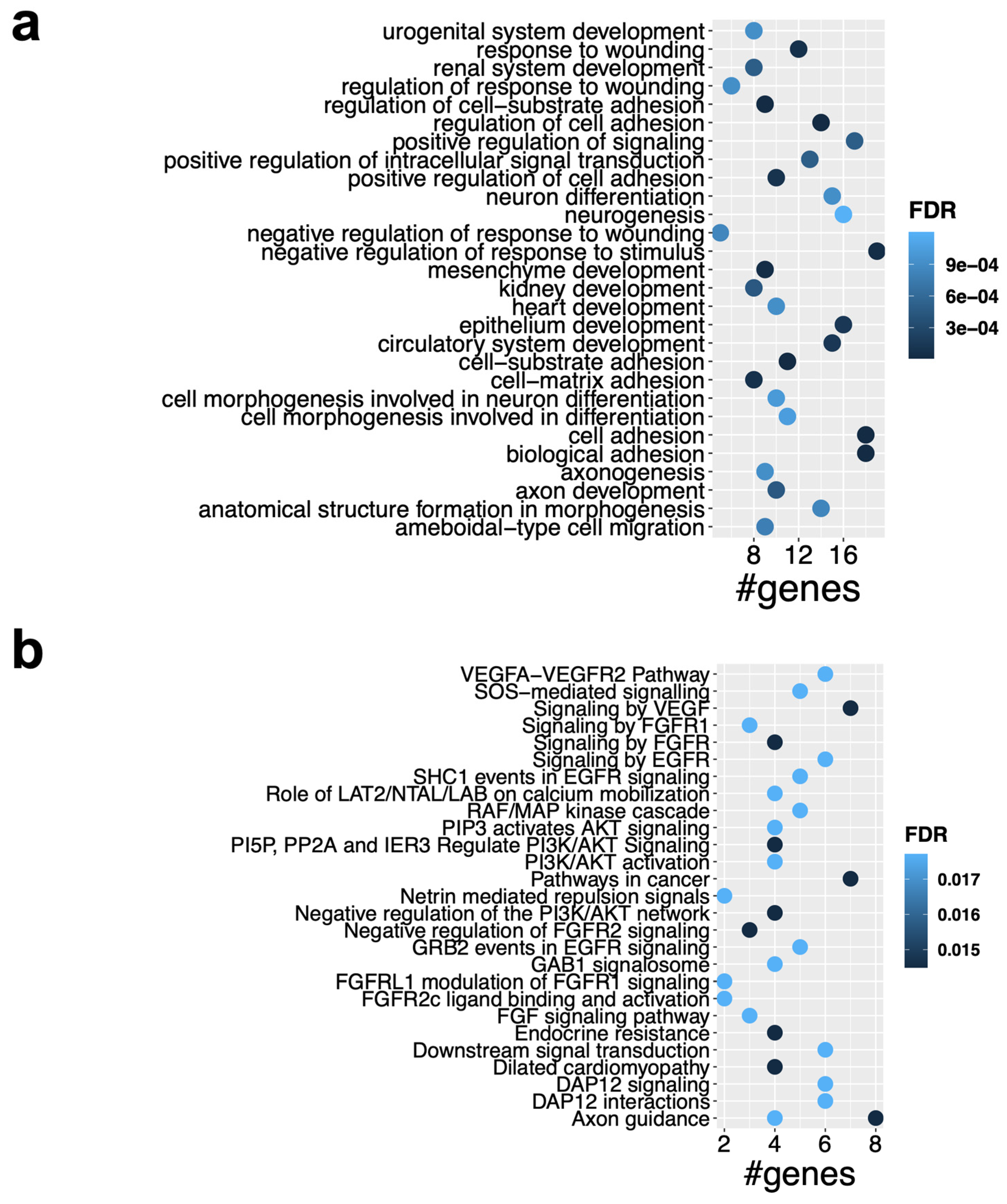
2.5. Functional Enrichment Analysis of the ceRNA Subnetworks
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.2. Inducing Oxidative Stress
4.3. RNA Extraction and Integrity Check
4.4. Total RNA-Seq Library Generation and Sequencing
4.5. Small RNA Sequencing
4.6. CircRNA Detection and Expression Analysis
4.7. cDNA Synthesis and Quantitative PCR
4.8. mRNA and miRNA Expression Analysis
4.9. Co-Expression Analysis
4.10. Construction of circRNA–miRNA–mRNA Network
4.11. Functional Enrichment Analysis
4.12. Visualization
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef]
- Salim, S. Oxidative stress and psychological disorders. Curr. Neuropharmacol. 2014, 12, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxid. Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef] [PubMed]
- Boskovic, M.; Vovk, T.; Kores Plesnicar, B.; Grabnar, I. Oxidative stress in schizophrenia. Curr. Neuropharmacol. 2011, 9, 301–312. [Google Scholar] [PubMed]
- Andreazza, A.C.; Kauer-Sant'anna, M.; Frey, B.N.; Bond, D.J.; Kapczinski, F.; Young, L.T.; Yatham, L.N. Oxidative stress markers in bipolar disorder: A meta-analysis. J. Affect. Disord. 2008, 111, 135–144. [Google Scholar] [CrossRef]
- Bajpai, A.; Verma, A.K.; Srivastava, M.; Srivastava, R. Oxidative stress and major depression. J. Clin. Diagn. Res. 2014, 8, CC04–CC07. [Google Scholar] [CrossRef]
- Pandya, C.D.; Howell, K.R.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 214–223. [Google Scholar] [CrossRef]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef]
- Kunsch, C.; Medford, R.M. Oxidative stress as a regulator of gene expression in the vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Han, E.S.; Muller, F.L.; Perez, V.I.; Qi, W.; Liang, H.; Xi, L.; Fu, C.; Doyle, E.; Hickey, M.; Cornell, J.; et al. The in vivo gene expression signature of oxidative stress. Physiol. Genom. 2008, 34, 112–126. [Google Scholar] [CrossRef]
- Khavari, B.; Mahmoudi, E.; Geaghan, M.P.; Cairns, M.J. Oxidative Stress Impact on the Transcriptome of Differentiating Neuroblastoma Cells: Implication for Psychiatric Disorders. Int. J. Mol. Sci. 2020, 21, 9182. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. Non-coding RNAs are inolved in the response to oxidative stress. Biomed. Pharmacother. 2020, 127, 110228. [Google Scholar] [CrossRef]
- Wang, W.-T.; Ye, H.; Wei, P.-P.; Han, B.-W.; He, B.; Chen, Z.-H.; Chen, Y.-Q. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J. Hematol. Oncol. 2016, 9, 117. [Google Scholar] [CrossRef]
- Khavari, B.; Michelle, M.B.; Mahmoudi, E.; Geaghan, M.P.; Graham, A.; Cairns, M.J. microRNA and the Post-Transcriptional Response to Oxidative Stress during Neuronal Differentiation: Implications for Neurodevelopmental and Psychiatric Disorders. Life 2024, 14, 562. [Google Scholar] [CrossRef]
- Huang, S.; Yang, B.; Chen, B.J.; Bliim, N.; Ueberham, U.; Arendt, T.; Janitz, M. The emerging role of circular RNAs in transcriptome regulation. Genomics 2017, 109, 401–407. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Cairns, M.J. Circular RNAs are temporospatially regulated throughout development and ageing in the rat. Sci. Rep. 2019, 9, 2564. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Kiltschewskij, D.; Fitzsimmons, C.; Cairns, M.J. Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation. Cells 2019, 9, 25. [Google Scholar] [CrossRef]
- Huang, A.; Zheng, H.; Wu, Z.; Chen, M.; Huang, Y. Circular RNA-protein interactions: Functions, mechanisms, and identification. Theranostics 2020, 10, 3503–3517. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Kuo, H.-C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef]
- Li, Z.; Cheng, Y.; Wu, F.; Wu, L.; Cao, H.; Wang, Q.; Tang, W. The emerging landscape of circular RNAs in immunity: Breakthroughs and challenges. Biomark. Res. 2020, 8, 25. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Fitzsimmons, C.; Geaghan, M.P.; Shannon Weickert, C.; Atkins, J.R.; Wang, X.; Cairns, M.J. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacology 2019, 44, 1043–1054. [Google Scholar] [CrossRef]
- Li, Z.; Liu, S.; Li, X.; Zhao, W.; Li, J.; Xu, Y. Circular RNA in Schizophrenia and Depression. Front. Psychiatry 2020, 11, 392. [Google Scholar] [CrossRef]
- Qu, S.; Zhong, Y.; Shang, R.; Zhang, X.; Song, W.; Kjems, J.; Li, H. The emerging landscape of circular RNA in life processes. RNA Biol. 2017, 14, 992–999. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 2019, 10, 503. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Piwecka, M.; Glazar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Yang, J.; Zhao, F. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat. Commun. 2020, 11, 90. [Google Scholar] [CrossRef]
- Smigielski, L.; Jagannath, V.; Rössler, W.; Walitza, S.; Grünblatt, E. Epigenetic mechanisms in schizophrenia and other psychotic disorders: A systematic review of empirical human findings. Mol. Psychiatry 2020, 25, 1718–1748. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gao, R.; Li, S.; Li, N.; Jiang, K.; Sun, X.; Zhang, J. Circular RNA circZNF292 regulates H2 O2 -induced injury in human lens epithelial HLE-B3 cells depending on the regulation of the miR-222-3p/E2F3 axis. Cell Biol. Int. 2021, 45, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Di Liddo, A.; de Oliveira Freitas Machado, C.; Fischer, S.; Ebersberger, S.; Heumüller, A.W.; Weigand, J.E.; Müller-McNicoll, M.; Zarnack, K. A combined computational pipeline to detect circular RNAs in human cancer cells under hypoxic stress. J. Mol. Cell. Biol. 2019, 11, 829–844. [Google Scholar] [CrossRef]
- Boeckel, J.N.; Jaé, N.; Heumüller, A.W.; Chen, W.; Boon, R.A.; Stellos, K.; Zeiher, A.M.; John, D.; Uchida, S.; Dimmeler, S. Identification and Characterization of Hypoxia-Regulated Endothelial Circular RNA. Circ. Res. 2015, 117, 884–890. [Google Scholar] [CrossRef]
- Bi, J.; Liu, H.; Cai, Z.; Dong, W.; Jiang, N.; Yang, M.; Huang, J.; Lin, T. Circ-BPTF promotes bladder cancer progression and recurrence through the miR-31-5p/RAB27A axis. Aging 2018, 10, 1964–1976. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, N.; Zhang, S.; Zhou, P.; Lv, L.; Richard, S.A.; Ma, W.; Chen, C.; Wang, X.; Huang, S.; et al. Differential circular RNA expression profiles of invasive and non-invasive non-functioning pituitary adenomas: A microarray analysis. Medicine 2019, 98, e16148. [Google Scholar] [CrossRef]
- Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Cho, S.G. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Wilkinson, E.; Cui, Y.H.; He, Y.Y. Context-Dependent Roles of RNA Modifications in Stress Responses and Diseases. Int. J. Mol. Sci. 2021, 22, 1949. [Google Scholar] [CrossRef]
- Luo, X.; Sun, D.; Wang, Y.; Zhang, F.; Wang, Y. Cpt1a promoted ROS-induced oxidative stress and inflammation in liver injury via the Nrf2/HO-1 and NLRP3 inflammasome signaling pathway. Can. J. Physiol. Pharmacol. 2021, 99, 468–477. [Google Scholar] [CrossRef]
- Joshi, M.; Kim, J.; D'Alessandro, A.; Monk, E.; Bruce, K.; Elajaili, H.; Nozik-Grayck, E.; Goodspeed, A.; Costello, J.C.; Schlaepfer, I.R. CPT1A Over-Expression Increases Reactive Oxygen Species in the Mitochondria and Promotes Antioxidant Defenses in Prostate Cancer. Cancers 2020, 12, 3431. [Google Scholar] [CrossRef]
- Gao, X.; Ma, X.K.; Li, X.; Li, G.W.; Liu, C.X.; Zhang, J.; Wang, Y.; Wei, J.; Chen, J.; Chen, L.L.; et al. Knockout of circRNAs by base editing back-splice sites of circularized exons. Genome Biol. 2022, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Pisignano, G.; Michael, D.C.; Visal, T.H.; Pirlog, R.; Ladomery, M.; Calin, G.A. Going circular: History, present, and future of circRNAs in cancer. Oncogene 2023, 42, 2783–2800. [Google Scholar] [CrossRef] [PubMed]
- Di Liegro, C.M.; Schiera, G.; Schirò, G.; Di Liegro, I. RNA-Binding Proteins as Epigenetic Regulators of Brain Functions and Their Involvement in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 14622. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Liu, H.; Ma, J.; Yuan, J.; Li, X.; Qiao, L. A Narrative Review of Circular RNAs in Brain Development and Diseases of Preterm Infants. Front. Pediatr. 2021, 21, 706012. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Chandra, K.; Al-Harthi, S.; Sukumaran, S.; Almulhim, F.; Emwas, A.H.; Atreya, H.S.; Jaremko, Ł.; Jaremko, M. NMR-based metabolomics with enhanced sensitivity. RSC Adv. 2021, 11, 8694–8700. [Google Scholar] [CrossRef]
- Chandra, K.; Al-Harthi, S.; Almulhim, F.; Emwas, A.H.; Jaremko, Ł.; Jaremko, M. The robust NMR toolbox for metabolomics. Mol. Omics 2021, 17, 719–724. [Google Scholar] [CrossRef]
- Hameister, R.; Kaur, C.; Dheen, S.T.; Lohmann, C.H.; Singh, G. Reactive oxygen/nitrogen species (ROS/RNS) and oxidative stress in arthroplasty. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 2073–2087. [Google Scholar] [CrossRef]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry. 2015, 20, 361–368. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Pfrieger, F.W. The RIN: An RNA integrity number for the assessment of RNA quality. Nat. Protoc. 2006, 1, 1044–1050. [Google Scholar]
- Chen, S.; Huang, T.; Zhou, Y.; Han, Y.; Xu, M.; Gu, J. AfterQC: Automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinform. 2017, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html (accessed on 6 November 2024).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs Sponged by circRNAs | miRNAs Not Sponged by circRNAs | Total | |
|---|---|---|---|
| miRNAs associated with psychiatry | circRNA-sponged, Psy-associated miRNAs 66 | Not sponged, Psy-associated miRNAs 105 | Total number of Psy-associated miRNAs 171 |
| miRNAs unassociated with psychiatry | circRNA-sponged, Psy-unassociated miRNAs 48 | Not sponged, Psy-unassociated miRNAs 665 | Total number of Psy-unassociated miRNAs 713 |
| Total | Total number of circRNA-sponged miRNAs 114 | Total number of miRNAs not sponged 770 | Total number of miRNAs expressed in the cell line 884 |
| miRNAs Sponged by circRNAs | miRNAs Not Sponged by circRNAs | Total | |
|---|---|---|---|
| miRNAs associated with psychiatry | circRNA-sponged, Psy-associated miRNAs 36 | Not sponged, Psy-associated miRNAs 130 | Total number of Psy-associated miRNAs 166 |
| miRNAs unassociated with psychiatry | circRNA-sponged, Psy-unassociated miRNAs 24 | Not sponged, Psy-unassociated miRNAs 612 | Total number of Psy-unassociated miRNAs 636 |
| Total | Total number of circRNA-sponged miRNAs 60 | Total number of miRNAs not sponged 742 | Total number of miRNAs expressed in the cell line 802 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoudi, E.; Khavari, B.; Cairns, M.J. Oxidative Stress-Associated Alteration of circRNA and Their ceRNA Network in Differentiating Neuroblasts. Int. J. Mol. Sci. 2024, 25, 12459. https://doi.org/10.3390/ijms252212459
Mahmoudi E, Khavari B, Cairns MJ. Oxidative Stress-Associated Alteration of circRNA and Their ceRNA Network in Differentiating Neuroblasts. International Journal of Molecular Sciences. 2024; 25(22):12459. https://doi.org/10.3390/ijms252212459
Chicago/Turabian StyleMahmoudi, Ebrahim, Behnaz Khavari, and Murray J. Cairns. 2024. "Oxidative Stress-Associated Alteration of circRNA and Their ceRNA Network in Differentiating Neuroblasts" International Journal of Molecular Sciences 25, no. 22: 12459. https://doi.org/10.3390/ijms252212459
APA StyleMahmoudi, E., Khavari, B., & Cairns, M. J. (2024). Oxidative Stress-Associated Alteration of circRNA and Their ceRNA Network in Differentiating Neuroblasts. International Journal of Molecular Sciences, 25(22), 12459. https://doi.org/10.3390/ijms252212459