
FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models
 , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
2. Results
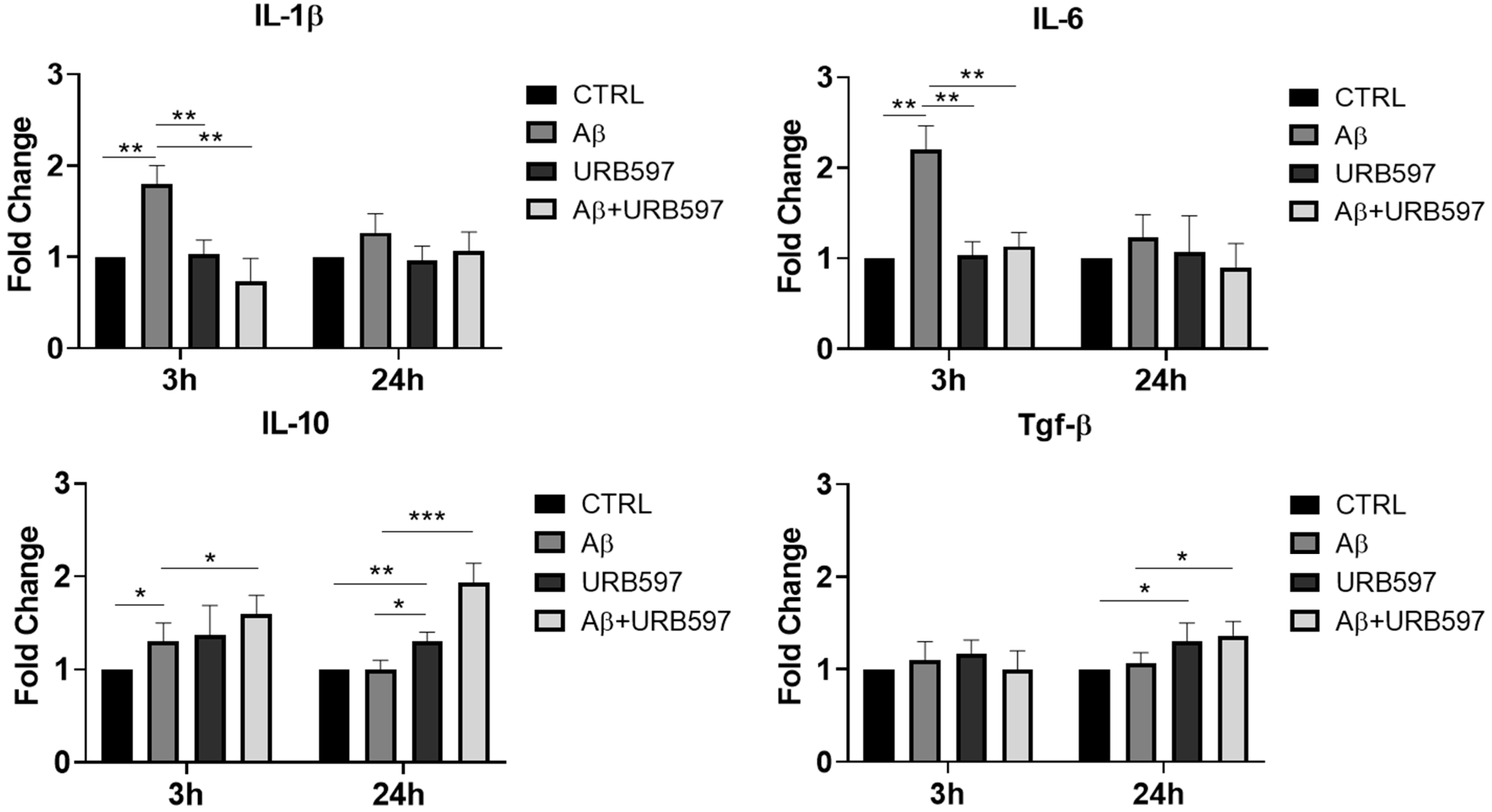
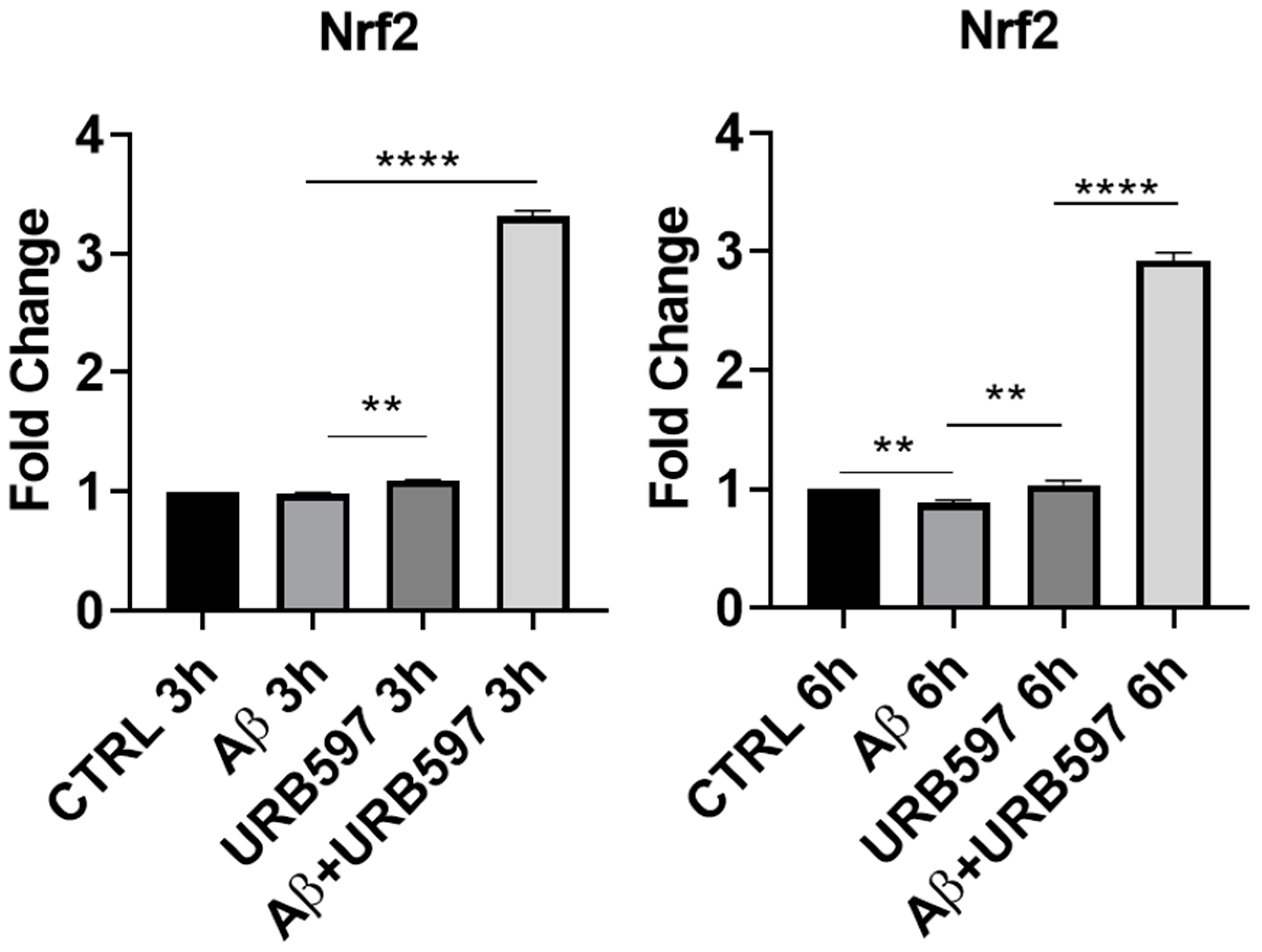
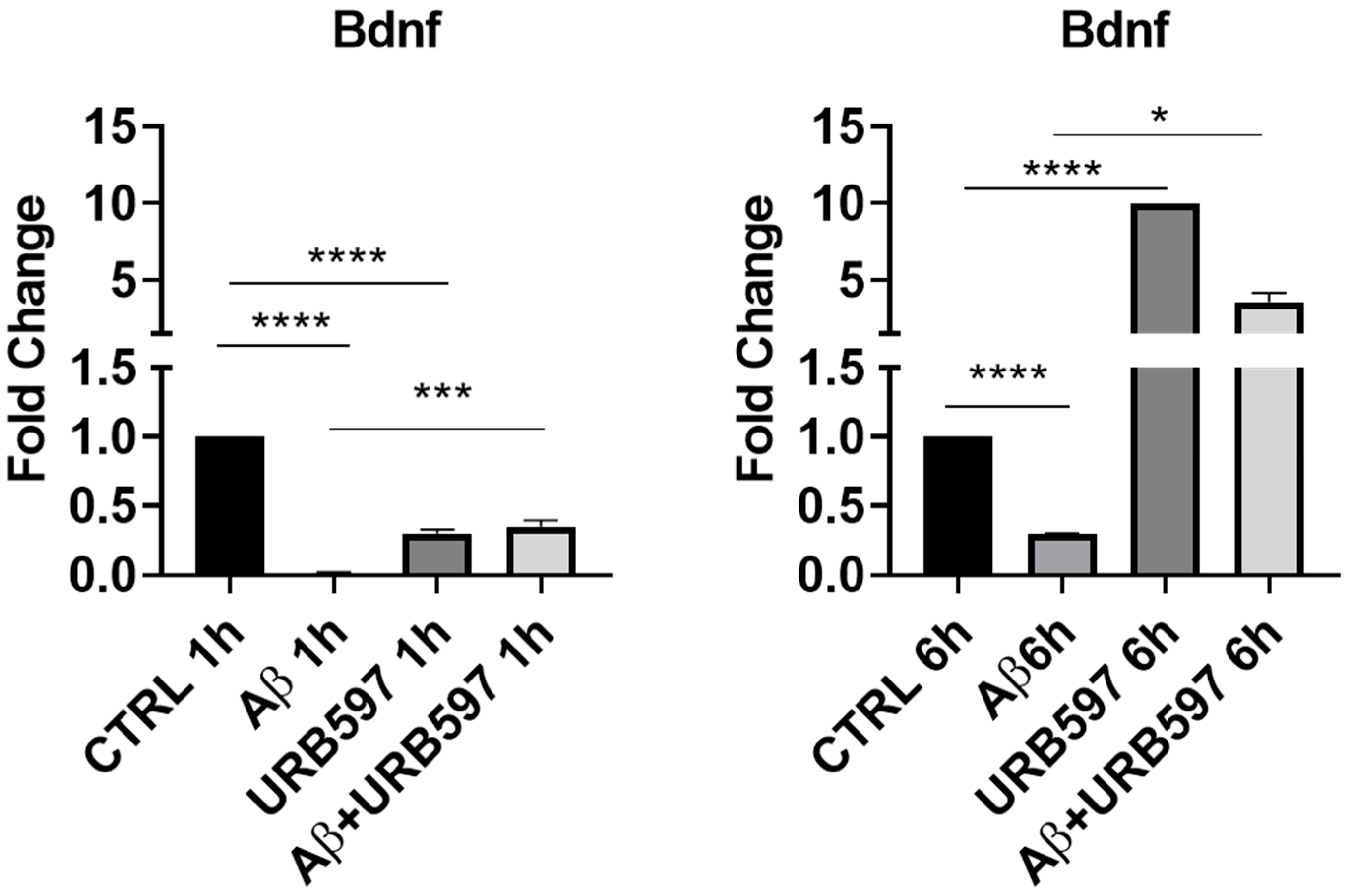
2.1. URB597 Treatment in BV-2 Cells
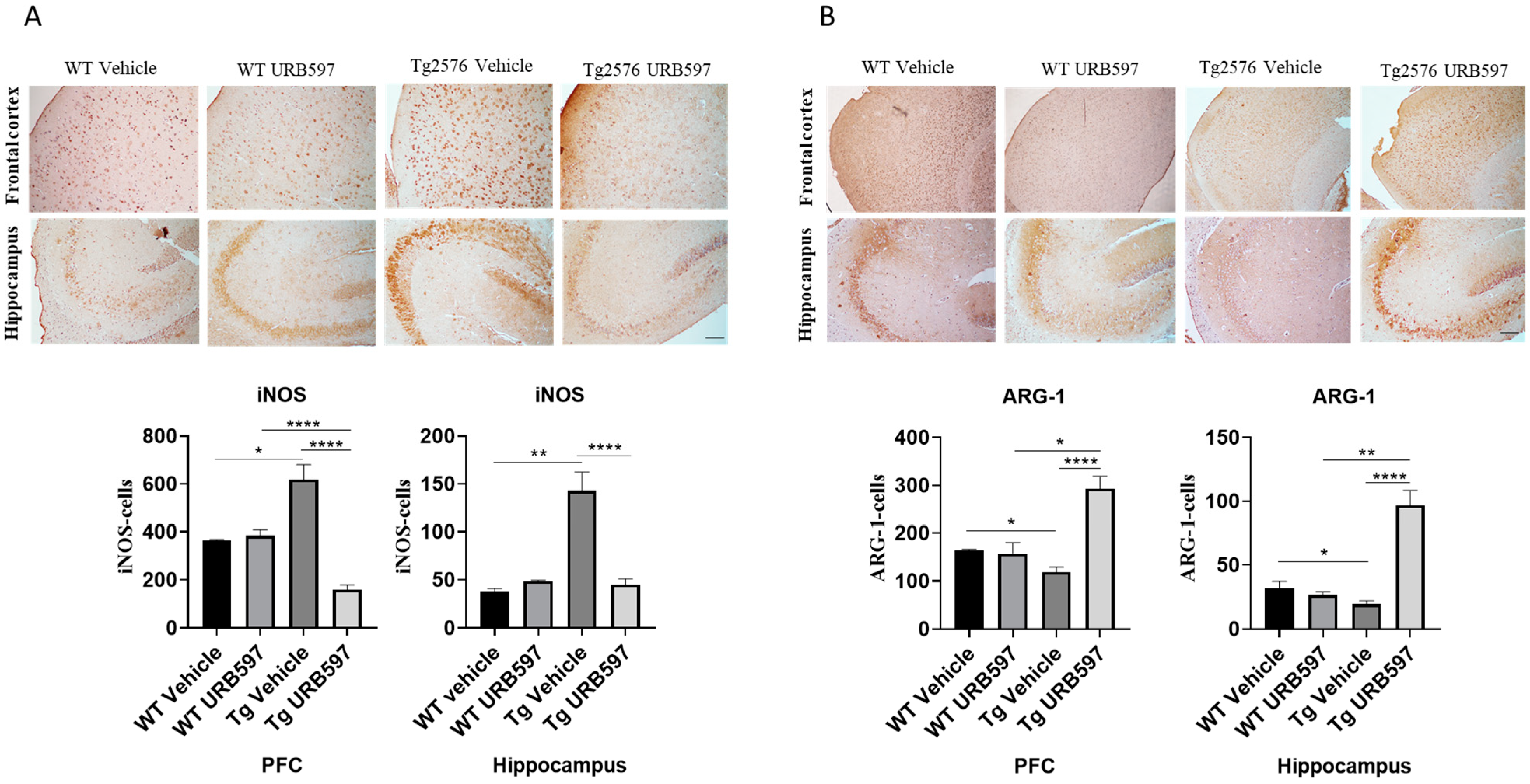
2.2. FAAH Inhibition in AD-like Mice
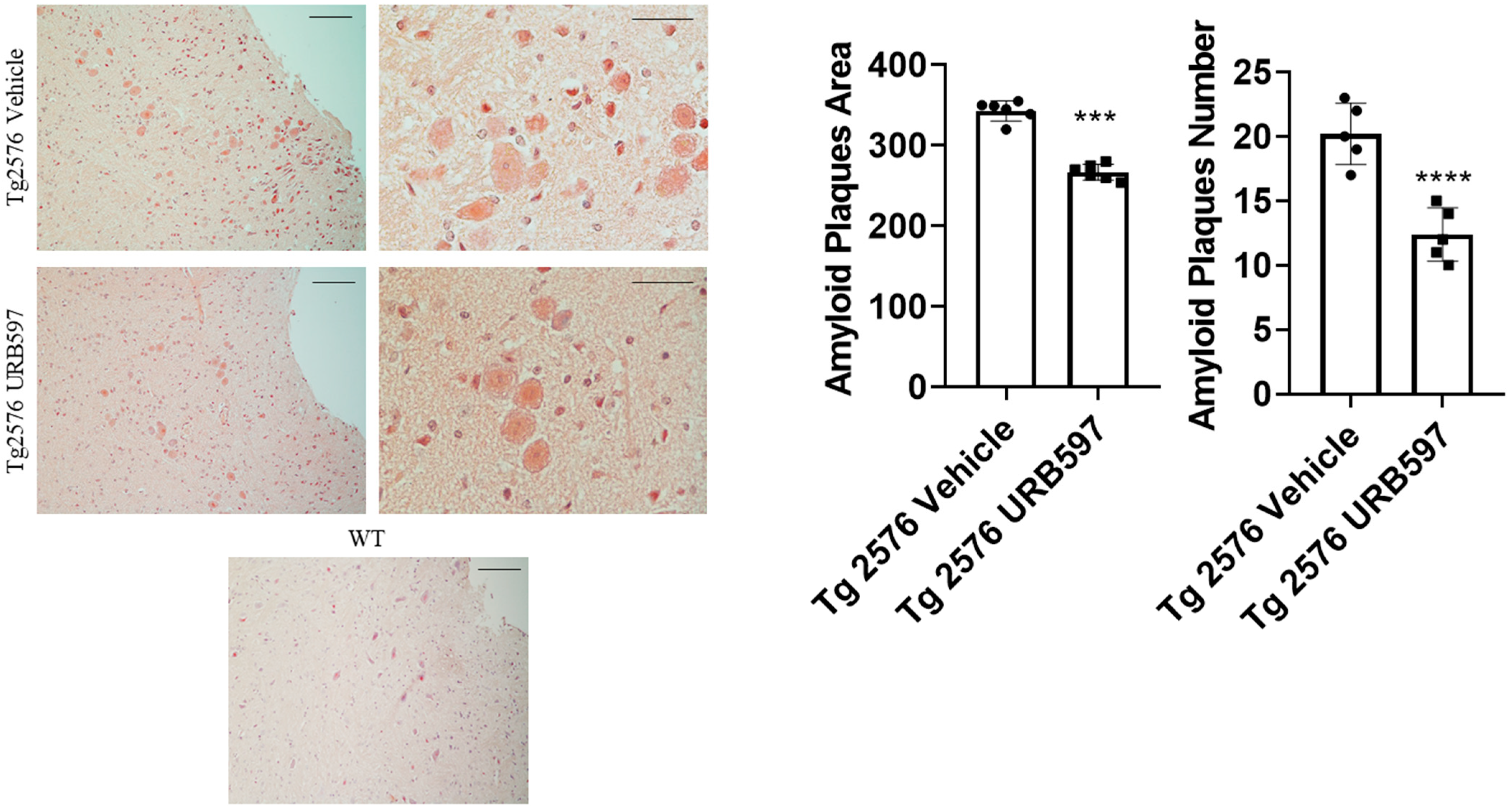
2.3. β-Amyloid Aggregation
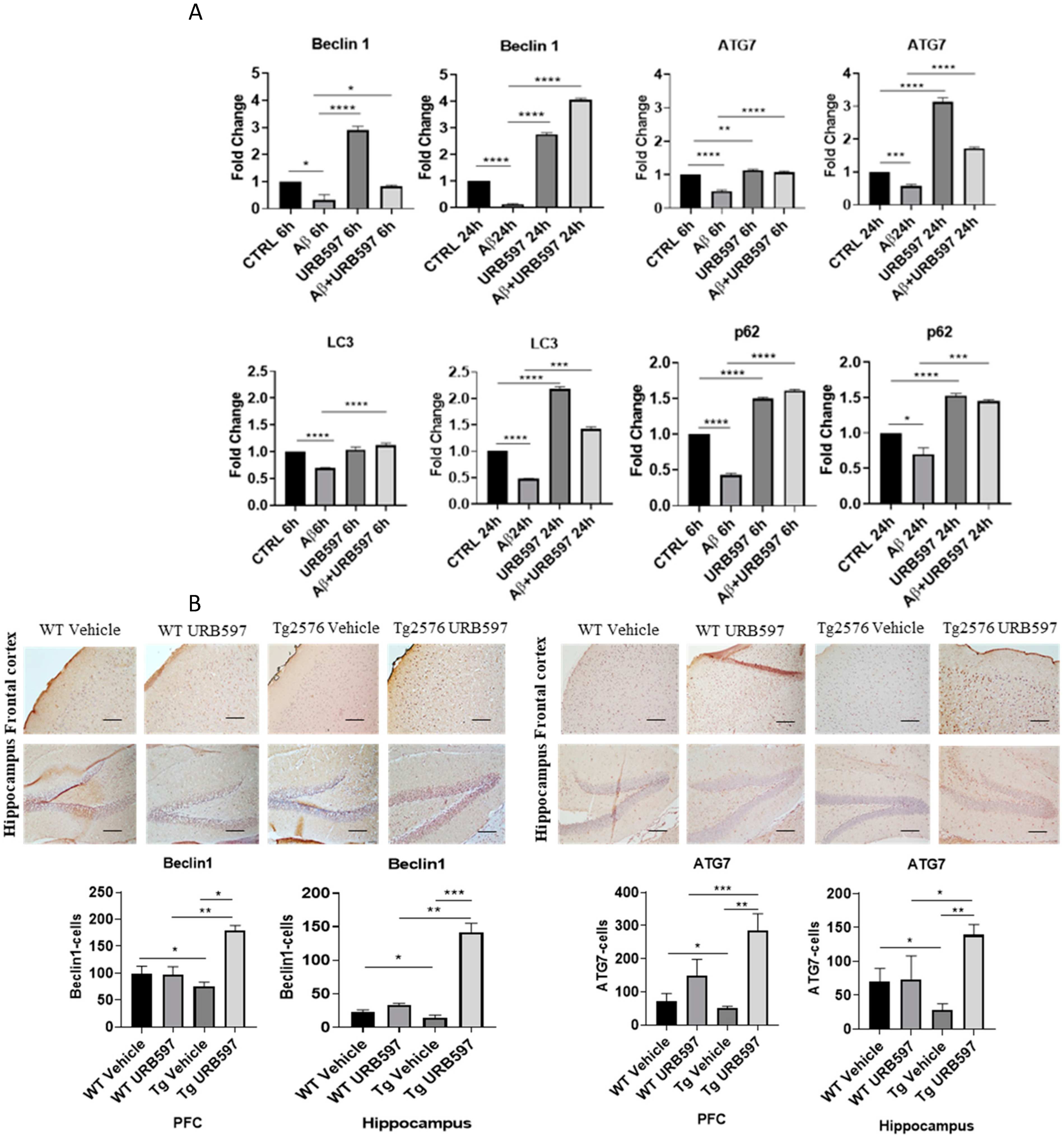
2.4. URB597 and Autophagy
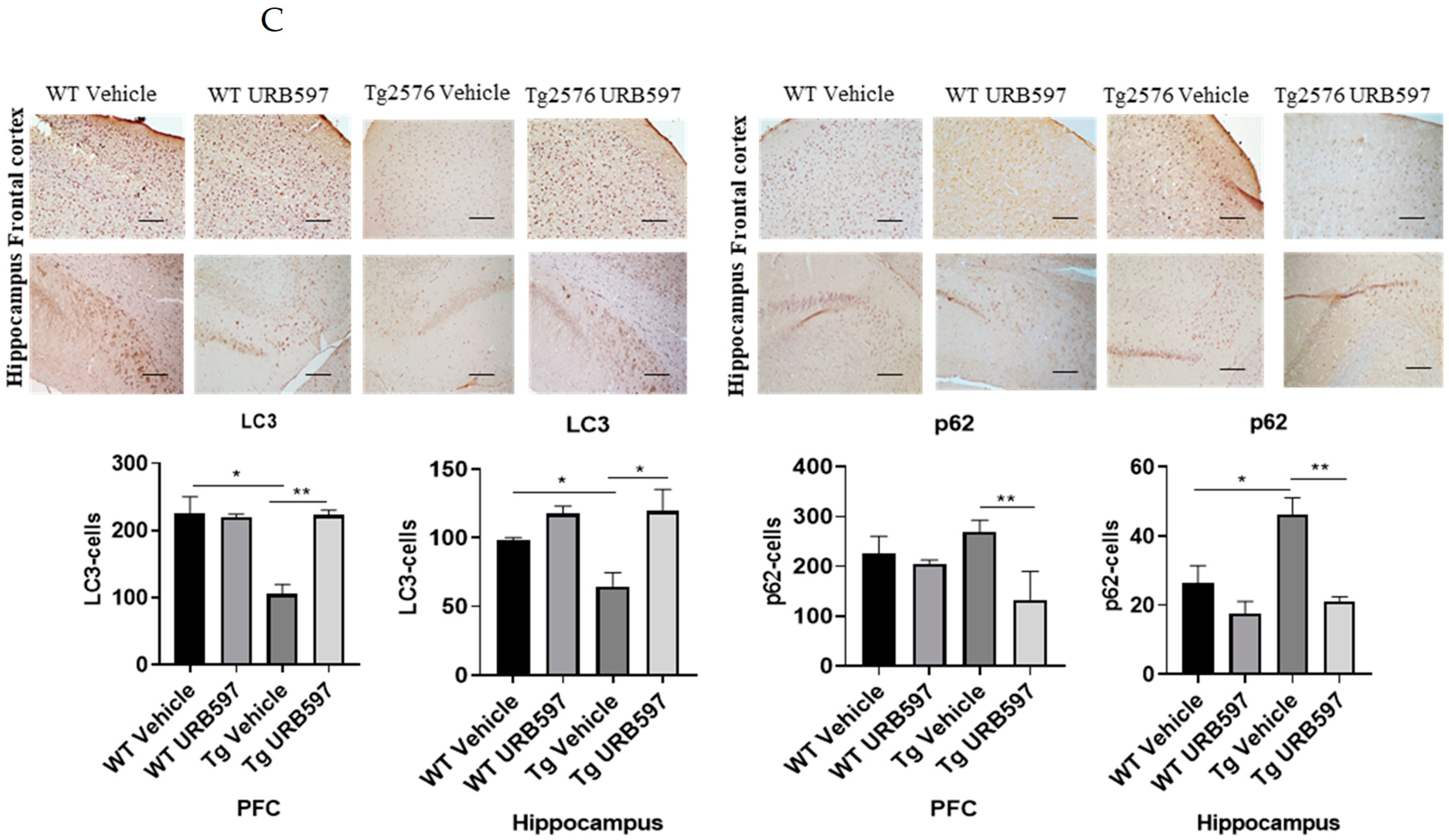
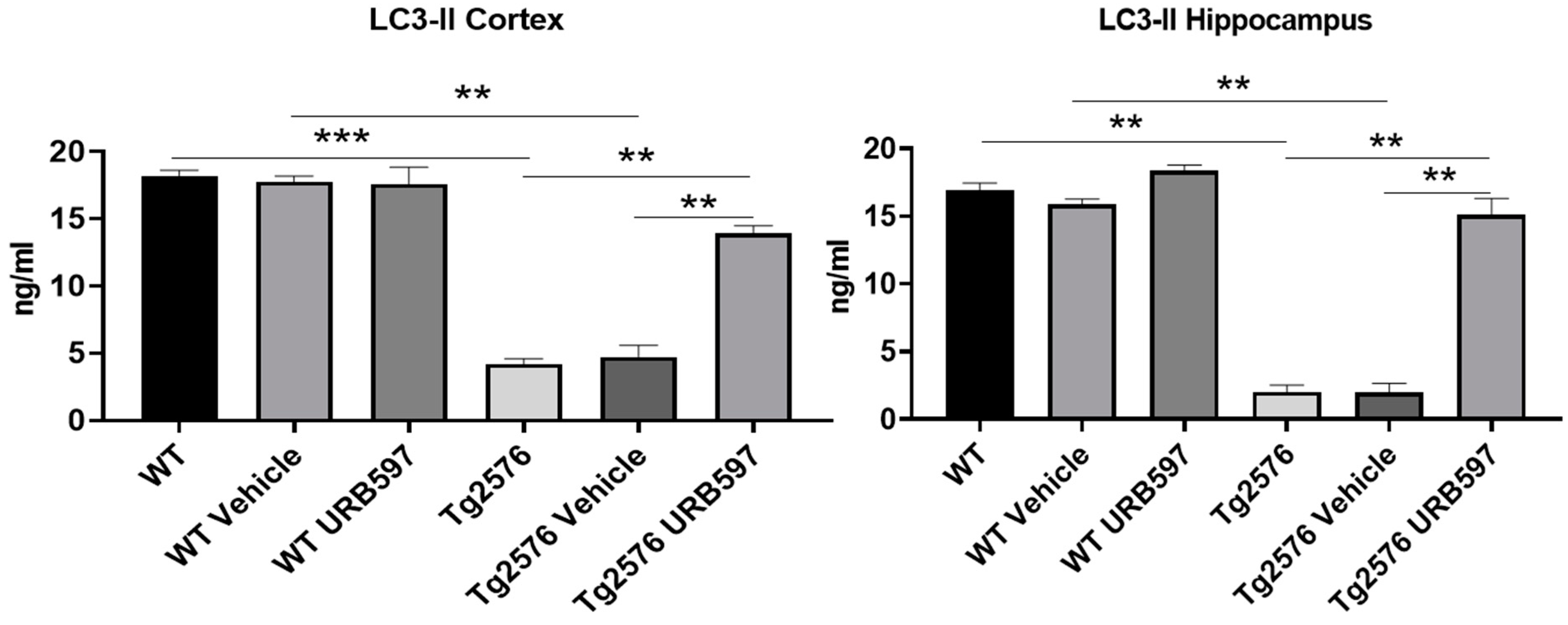
2.5. FAAH Inhibition: LC3-II Expression in AD-like Mice
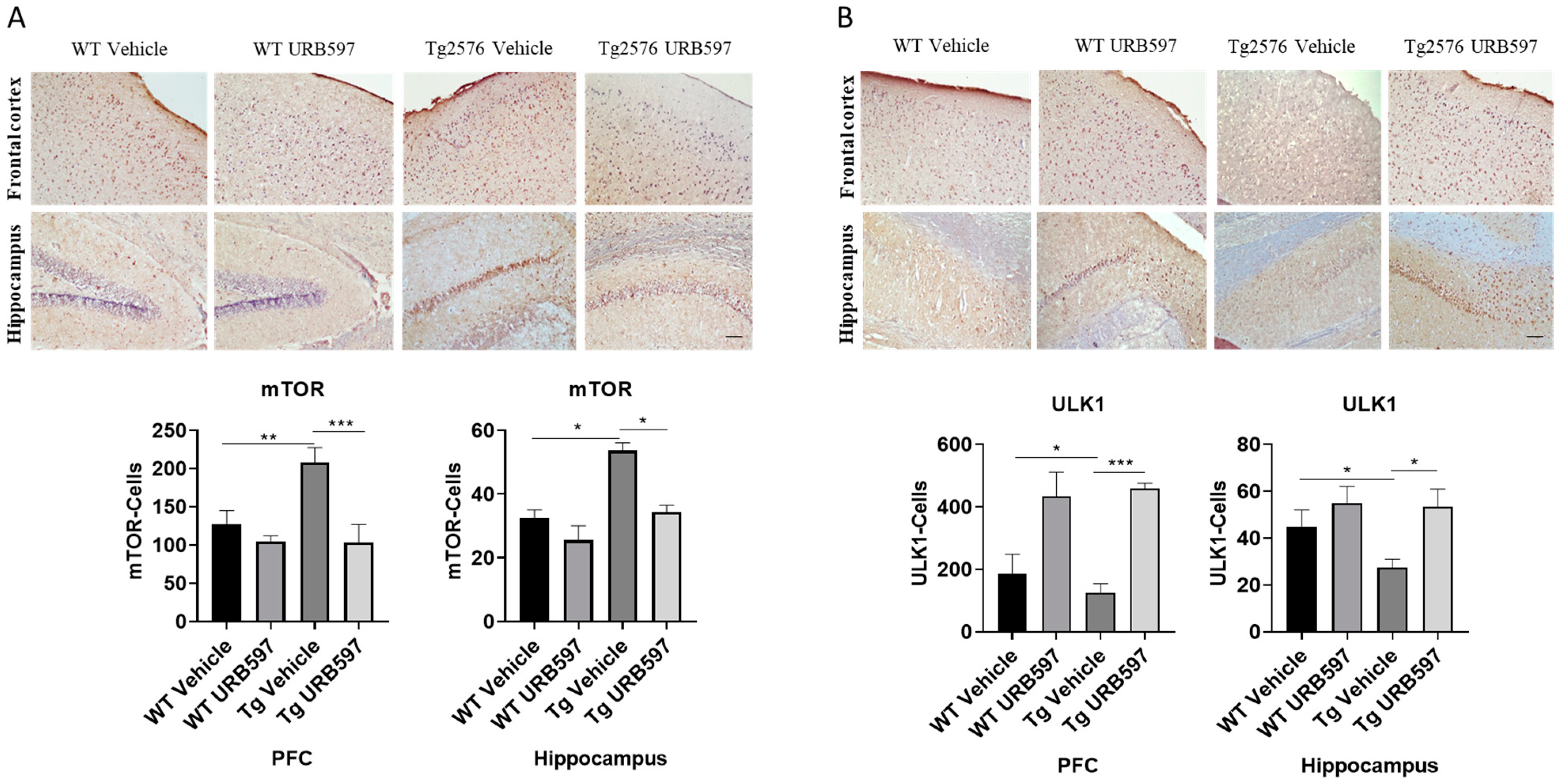
2.6. mTOR and ULK1 Expression in AD-like Mice
3. Discussion
4. Limitations
5. Materials and Methods
5.1. In Vivo Experimental Design
5.2. In Vitro Analysis
5.3. Cell Cultures and Treatments
5.4. Real-Time Quantitative PCR Analysis
5.5. Immunohistochemistry
5.6. Congo Red Method
5.7. ELISA Assay
5.8. Statistics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| AEA | anandamide |
| ARG-1 | arginase-1 |
| ATG7 | autophagy-related 7 |
| BDNF | brain-derived neurotrophic factor |
| BECN1 | Beclin 1 |
| CB1 | cannabinoid type-1 receptors |
| CB2 | cannabinoid type-2 receptors |
| CTRL | control |
| FAAH | fatty acid amide hydrolase |
| eCBs | endocannabinoids |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IL-10 | Interleukin 10 |
| iNOS | inducible nitric oxide synthase |
| mTOR | mammalian target of rapamycin |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PFC | prefrontal cortex |
| ROS | reactive oxygen species |
| SQSTM1/p62 | sequestosome 1 |
| TGF-β | transforming growth factor-beta |
| TNF-α | tumor necrosis factor alpha |
References
- Maccarrone, M.; Di Marzo, V.; Gertsch, J.; Grether, U.; Howlett, A.C.; Hua, T.; Makriyannis, A.; Piomelli, D.; Ueda, N.; Van Der Stelt, M. Goods and Bads of the Endocannabinoid System as a Therapeutic Target: Lessons Learned after 30 Years. Pharmacol. Rev. 2023, 75, 885–958. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular Characterization of an Enzyme That Degrades Neuromodulatory Fatty-Acid Amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Komarnytsky, S.; Rathinasabapathy, T.; Wagner, C.; Metzger, B.; Carlisle, C.; Panda, C.; Le Brun-Blashka, S.; Troup, J.P.; Varadharaj, S. Endocannabinoid System and Its Regulation by Polyunsaturated Fatty Acids and Full Spectrum Hemp Oils. Int. J. Mol. Sci. 2021, 22, 5479. [Google Scholar] [CrossRef]
- Chen, C. Homeostatic Regulation of Brain Functions by Endocannabinoid Signaling. Neural Regen. Res. 2015, 10, 691. [Google Scholar] [CrossRef]
- Chen, C. Endocannabinoid Metabolism and Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 1987. [Google Scholar] [CrossRef]
- McKenna, M.; McDougall, J.J. Cannabinoid Control of Neurogenic Inflammation. Br. J. Pharmacol. 2020, 177, 4386–4399. [Google Scholar] [CrossRef]
- Grieco, M.; De Caris, M.G.; Maggi, E.; Armeli, F.; Coccurello, R.; Bisogno, T.; D’Erme, M.; Maccarrone, M.; Mancini, P.; Businaro, R. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. Int. J. Mol. Sci. 2021, 22, 7711. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.D.; Landreth, G.E. The Role of Microglia in Amyloid Clearance from the AD Brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Taneo, J.; Adachi, T.; Yoshida, A.; Takayasu, K.; Takahara, K.; Inaba, K. Amyloid β Oligomers Induce Interleukin-1β Production in Primary Microglia in a Cathepsin B- and Reactive Oxygen Species-Dependent Manner. Biochem. Biophys. Res. Commun. 2015, 458, 561–567. [Google Scholar] [CrossRef]
- Griffin, W.S.T.; Mrak, R.E. Interleukin-1 in the Genesis and Progression of and Risk for Development of Neuronal Degeneration in Alzheimer’s Disease. J. Leukoc. Biol. 2002, 72, 233–238. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial Activation and Amyloid Deposition in Mild Cognitive Impairment: A PET Study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef]
- Scipioni, L.; Ciaramellano, F.; Carnicelli, V.; Leuti, A.; Lizzi, A.R.; De Dominicis, N.; Oddi, S.; Maccarrone, M. Microglial Endocannabinoid Signalling in AD. Cells 2022, 11, 1237. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured Rat Microglial Cells Synthesize the Endocannabinoid 2-Arachidonylglycerol, Which Increases Proliferation via a CB2 Receptor-Dependent Mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active Endocannabinoids Are Secreted on Extracellular Membrane Vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; López Rodríguez, M.L.; Bajjalieh, S.; Stella, N. Identification of a Novel Endocannabinoid-Hydrolyzing Enzyme Expressed by Microglial Cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Stella, N. Cannabinoid and Cannabinoid-like Receptors in Microglia, Astrocytes, and Astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yagyu, K.; Sackett, S.; Zhang, Y. Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 2019, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pérez, G.; Ruiz De Martín Esteban, S.; Marqués, S.; Aparicio, N.; Grande, M.T.; Benito-Cuesta, I.; Martínez-Relimpio, A.M.; Arnanz, M.A.; Tolón, R.M.; Posada-Ayala, M.; et al. Potentiation of Amyloid Beta Phagocytosis and Amelioration of Synaptic Dysfunction upon FAAH Deletion in a Mouse Model of Alzheimer’s Disease. J. Neuroinflamm. 2021, 18, 223. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 04. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, C.; Jiang, H. Novel Target for Treating Alzheimer’s Diseases: Crosstalk between the Nrf2 Pathway and Autophagy. Ageing Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.-Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A Specific Amyloid-Beta Protein Assembly in the Brain Impairs Memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Orta-Salazar, E.; Cuellar-Lemus, C.A.; Díaz-Cintra, S.; Feria-Velasco, A.I. Cholinergic Markers in the Cortex and Hippocampus of Some Animal Species and Their Correlation to Alzheimer’s Disease. Neurologia 2014, 29, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P.J. Untangling Autophagy Measurements: All Fluxed Up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular Neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [PubMed]
- Davoody, S.; Asgari Taei, A.; Khodabakhsh, P.; Dargahi, L. mTOR Signaling and Alzheimer’s Disease: What We Know and Where We Are? CNS Neurosci. Ther. 2024, 30, e14463. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Huang, W.; Zhong, Y.; Gao, B.; Zheng, B.; Liu, Y. Nrf2-Mediated Therapeutic Effects of Dietary Flavones in Different Diseases. Front. Pharmacol. 2023, 14, 1240433. [Google Scholar] [CrossRef]
- Yan, R.; Lin, B.; Jin, W.; Tang, L.; Hu, S.; Cai, R. NRF2, a Superstar of Ferroptosis. Antioxidants 2023, 12, 1739. [Google Scholar] [CrossRef]
- Valvassori, S.S.; Arent, C.O.; Steckert, A.V.; Varela, R.B.; Jornada, L.K.; Tonin, P.T.; Budni, J.; Mariot, E.; Kapczinski, F.; Quevedo, J. Intracerebral Administration of BDNF Protects Rat Brain Against Oxidative Stress Induced by Ouabain in an Animal Model of Mania. Mol. Neurobiol. 2015, 52, 353–362. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Ugidos, I.F.; Pérez-Rodríguez, D.; Anuncibay-Soto, B.; Santos-Galdiano, M.; Font-Belmonte, E.; Gonzalo-Orden, J.M.; Fernández-López, A. Brain-derived Neurotrophic Factor Alleviates the Oxidative Stress Induced by Oxygen and Glucose Deprivation in an Ex Vivo Brain Slice Model. J. Cell. Physiol. 2019, 234, 9592–9604. [Google Scholar] [CrossRef]
- Tolón, R.M.; Núñez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martínez-Orgado, J.A.; Romero, J. The Activation of Cannabinoid CB2 Receptors Stimulates in Situ and In Vitro Beta-Amyloid Removal by Human Macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Kumar, A.; Schliebs, R. Impairment of Cholinergic Neurotransmission in Adult and Aged Transgenic Tg2576 Mouse Brain Expressing the Swedish Mutation of Human β-Amyloid Precursor Protein. Brain Res. 2002, 953, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s Disease Pathogenesis: Therapeutic Potential and Future Perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhou, Y.; Xiao, M.; Yan, L.-J.; He, W. Activation of mTOR: A Culprit of Alzheimer’s Disease? Neuropsychiatr. Dis. Treat. 2015, 11, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, J. Magnolol Improves Alzheimer’s Disease-like Pathologies and Cognitive Decline by Promoting Autophagy through Activation of the AMPK/mTOR/ULK1 Pathway. Biomed. Pharmacother. 2023, 161, 114473. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Passani, M.B.; Coccurello, R. The Endocannabinoid-Like Derivative Oleoylethanolamide at the Gut–Brain Interface: A “Lipid Way” to Control Energy Intake and Body Weight. In Cannabinoids in Health and Disease; Meccariello, R., Chianese, R., Eds.; InTech: London, UK, 2016; ISBN 978-953-51-2429-0. [Google Scholar]
- Luo, R.; Su, L.-Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.-X.; Zhang, D.-F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-Mediated Autophagy Reduces Alzheimer Disease-like Pathology and Cognitive Decline in a Murine Model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Ahmad, A.; Bruschetta, G.; Di Paola, R.; Crupi, R.; Paterniti, I.; Esposito, E.; Cuzzocrea, S. The Anti-Inflammatory Effects of Palmitoylethanolamide (PEA) on Endotoxin-Induced Uveitis in Rats. Eur. J. Pharmacol. 2015, 761, 28–35. [Google Scholar] [CrossRef]
- Wang, D.; Lin, Q.; Su, S.; Liu, K.; Wu, Y.; Hai, J. URB597 Improves Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion by Inhibiting mTOR-Dependent Autophagy. Neuroscience 2017, 344, 293–304. [Google Scholar] [CrossRef]
- Su, S.-H.; Wu, Y.-F.; Lin, Q.; Wang, D.-P.; Hai, J. URB597 Protects against NLRP3 Inflammasome Activation by Inhibiting Autophagy Dysfunction in a Rat Model of Chronic Cerebral Hypoperfusion. J. Neuroinflamm. 2019, 16, 260. [Google Scholar] [CrossRef]
- Hiebel, C.; Kromm, T.; Stark, M.; Behl, C. Cannabinoid Receptor 1 Modulates the Autophagic Flux Independent of mTOR- and BECLIN1-complex. J. Neurochem. 2014, 131, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Koay, L.C.; Rigby, R.J.; Wright, K.L. Cannabinoid-Induced Autophagy Regulates Suppressor of Cytokine Signaling-3 in Intestinal Epithelium. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G140–G148. [Google Scholar] [CrossRef] [PubMed]
- Casarejos, M.J.; Perucho, J.; Gomez, A.; Muñoz, M.P.; Fernandez-Estevez, M.; Sagredo, O.; Fernandez Ruiz, J.; Guzman, M.; de Yebenes, J.G.; Mena, M.A. Natural Cannabinoids Improve Dopamine Neurotransmission and Tau and Amyloid Pathology in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2013, 35, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, Í.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid Action Induces Autophagy-Mediated Cell Death through Stimulation of ER Stress in Human Glioma Cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 Receptors Regulate Neuronal Energy Metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Bilkei-Gorzo, A.; Nozaki, C.; Togo, A.; Nakamura, K.; Ohta, K.; Zimmer, A.; Asahi, T. Age-Dependent Alteration in Mitochondrial Dynamics and Autophagy in Hippocampal Neuron of Cannabinoid CB1 Receptor-Deficient Mice. Brain Res. Bull. 2020, 160, 40–49. [Google Scholar] [CrossRef]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-β Peptide Activates Cultured Astrocytes: Morphological Alterations, Cytokine Induction and Nitric Oxide Release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Asiimwe, N.; Yeo, S.G.; Kim, M.-S.; Jung, J.; Jeong, N.Y. Nitric Oxide: Exploring the Contextual Link with Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2016, 2016, 7205747. [Google Scholar] [CrossRef]
- Haas, J.; Storch-Hagenlocher, B.; Biessmann, A.; Wildemann, B. Inducible Nitric Oxide Synthase and Argininosuccinate Synthetase: Co-Induction in Brain Tissue of Patients with Alzheimer’s Dementia and Following Stimulation with β-Amyloid 1–42 In Vitro. Neurosci. Lett. 2002, 322, 121–125. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Scipioni, L.; Arosio, B.; Mari, D.; Oddi, S.; Maccarrone, M. Anti-Inflammatory Effects of Fatty Acid Amide Hydrolase Inhibition in Monocytes/Macrophages from Alzheimer’s Disease Patients. Biomolecules 2021, 11, 502. [Google Scholar] [CrossRef]
- Jung, K.-M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An Amyloid Β42-Dependent Deficit in Anandamide Mobilization Is Associated with Cognitive Dysfunction in Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K. Transgenic Mice Expressing Alzheimer Amyloid Precursor Proteins. Exp. Gerontol. 1998, 33, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Totaro, A.; Leuti, A.; Giacovazzo, G.; Scipioni, L.; Mango, D.; Coccurello, R.; Nisticò, R.; Oddi, S. Early Alteration of Distribution and Activity of Hippocampal Type-1 Cannabinoid Receptor in Alzheimer’s Disease-like Mice Overexpressing the Human Mutant Amyloid Precursor Protein. Pharmacol. Res. 2018, 130, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Giacovazzo, G.; Bisogno, T.; Piscitelli, F.; Verde, R.; Oddi, S.; Maccarrone, M.; Coccurello, R. Different Routes to Inhibit Fatty Acid Amide Hydrolase: Do All Roads Lead to the Same Place? Int. J. Mol. Sci. 2019, 20, 4503. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of Murine Microglial Cells by a V-Raf / v-Myc Carrying Retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Accession Numbers |
|---|---|---|---|
| mIL-1β | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG | NM_008361.4 |
| mIL-6 | CGGAGAGGAGACTTCACAGAGGA | TTTCCACGATTTCCCAGAGAACA | NM_001314054.1 |
| mTgf-β | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG | NM_011577.2 |
| mIL-10 | GCCCTTTGCTATGGTGTCCTTTC | TCCCTGGTTTCTCTTCCCAAGAC | NM_010548.2 |
| mR18s | AAATCAGTTATGGTTCCTTTGGTC | GCTCTAGAATTACCACAGTTATCCAA | M27358 |
| mLc3 | TTCTTCCTCCTGGTGAATGG | GTCTCCTGCGAGGCATAAAC | NM_026160 |
| mBeclin1 | CAGCCTCTGAAACTGGACACGA | CTCTCCTGAGTTAGCCTCTTCC | NM_019584 |
| mNrf2 | TCTGAGCCAGGACTACGACG | GAGGTGGTGGTGGTGTCTCTGC | NM_010902 |
| mp62 | CCTTGCCCTACAGCTGAGTC | CCACACTCTCCCCCACATTC | NM_001290769 |
| mAtg7 | CAATGAGATCTGGGAAGCCATAA | AGGTCAAGAGCAGAAACTTGTTGA | NM_001253717 |
| mBdnf | GTGTGACAGTATTAGCGAGTGG | GCAGCCTTCCTTGGTGTAAC | NM_007540 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armeli, F.; Coccurello, R.; Giacovazzo, G.; Mengoni, B.; Paoletti, I.; Oddi, S.; Maccarrone, M.; Businaro, R. FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models. Int. J. Mol. Sci. 2024, 25, 12044. https://doi.org/10.3390/ijms252212044
Armeli F, Coccurello R, Giacovazzo G, Mengoni B, Paoletti I, Oddi S, Maccarrone M, Businaro R. FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models. International Journal of Molecular Sciences. 2024; 25(22):12044. https://doi.org/10.3390/ijms252212044
Chicago/Turabian StyleArmeli, Federica, Roberto Coccurello, Giacomo Giacovazzo, Beatrice Mengoni, Ilaria Paoletti, Sergio Oddi, Mauro Maccarrone, and Rita Businaro. 2024. "FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models" International Journal of Molecular Sciences 25, no. 22: 12044. https://doi.org/10.3390/ijms252212044
APA StyleArmeli, F., Coccurello, R., Giacovazzo, G., Mengoni, B., Paoletti, I., Oddi, S., Maccarrone, M., & Businaro, R. (2024). FAAH Inhibition Counteracts Neuroinflammation via Autophagy Recovery in AD Models. International Journal of Molecular Sciences, 25(22), 12044. https://doi.org/10.3390/ijms252212044