
Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
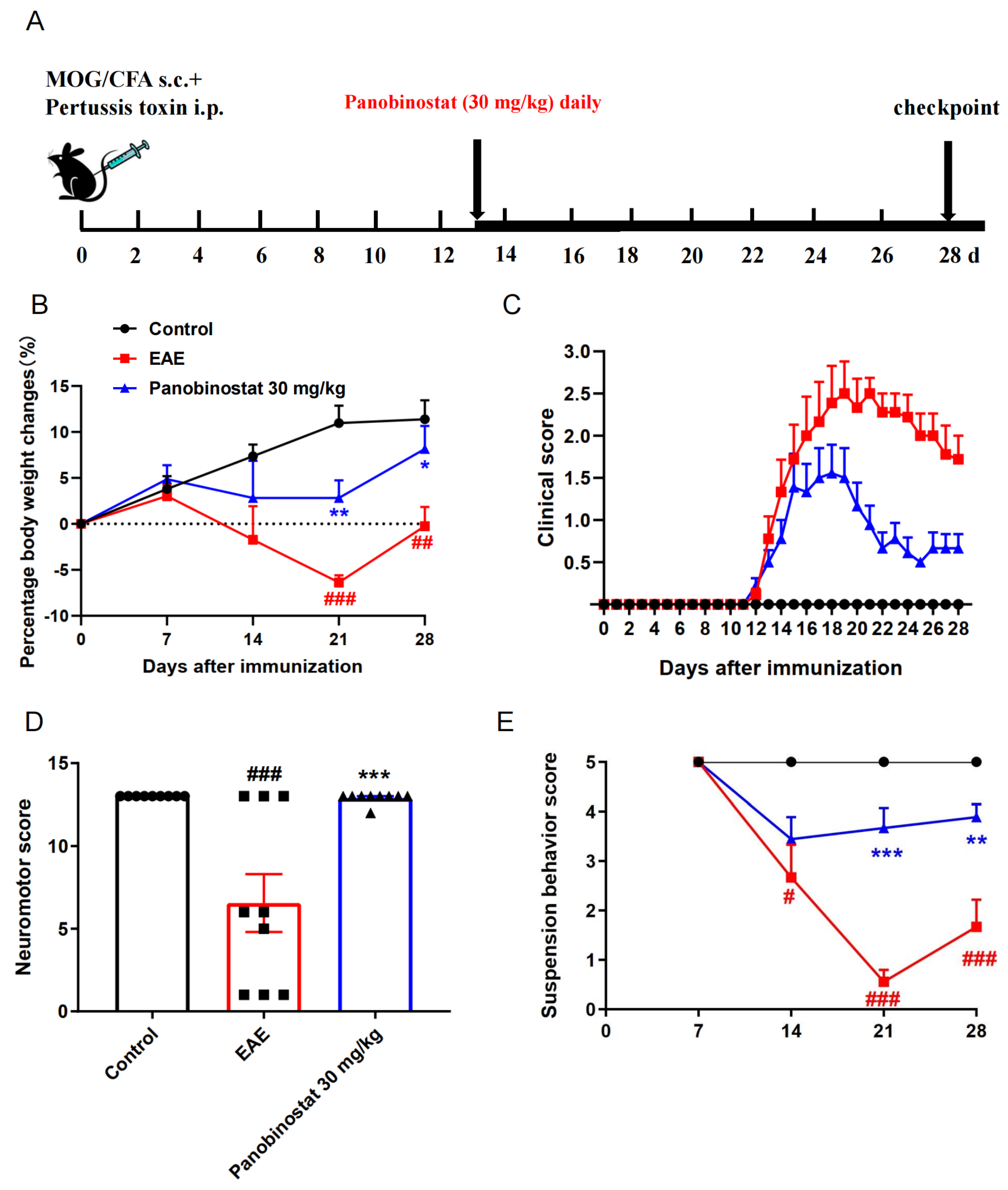
2.1. Panobinostat Alleviated EAE Symptoms in Mice
2.2. Panobinostat Ameliorated the CNS Demyelination and Loss of Oligodendrocytes in the Spinal Cords of EAE Mice
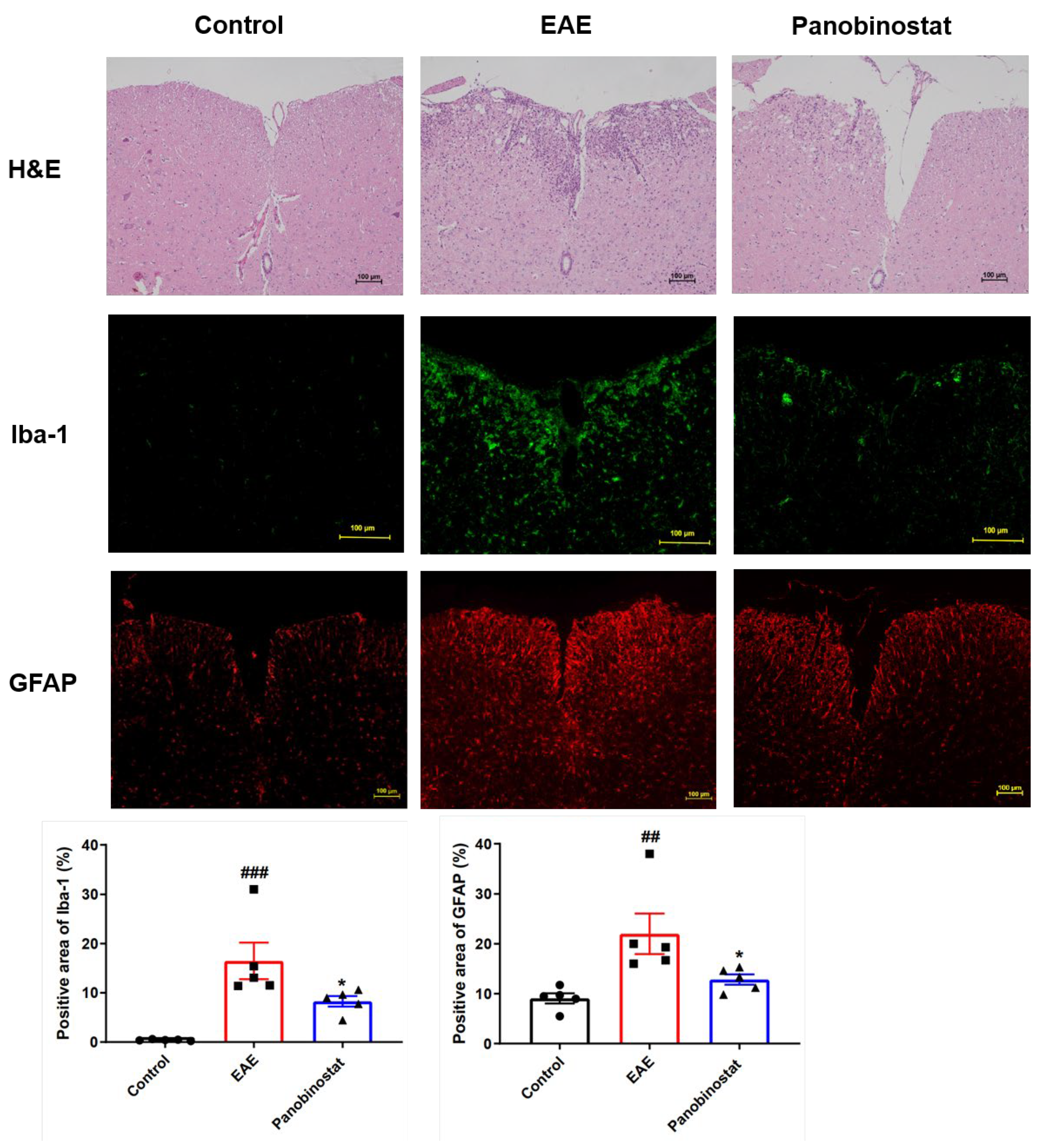
2.3. Panobinostat Diminished Inflammation and the Microglial/Astrogial Activation in the Spinal Cord Tissues of Mice with EAE
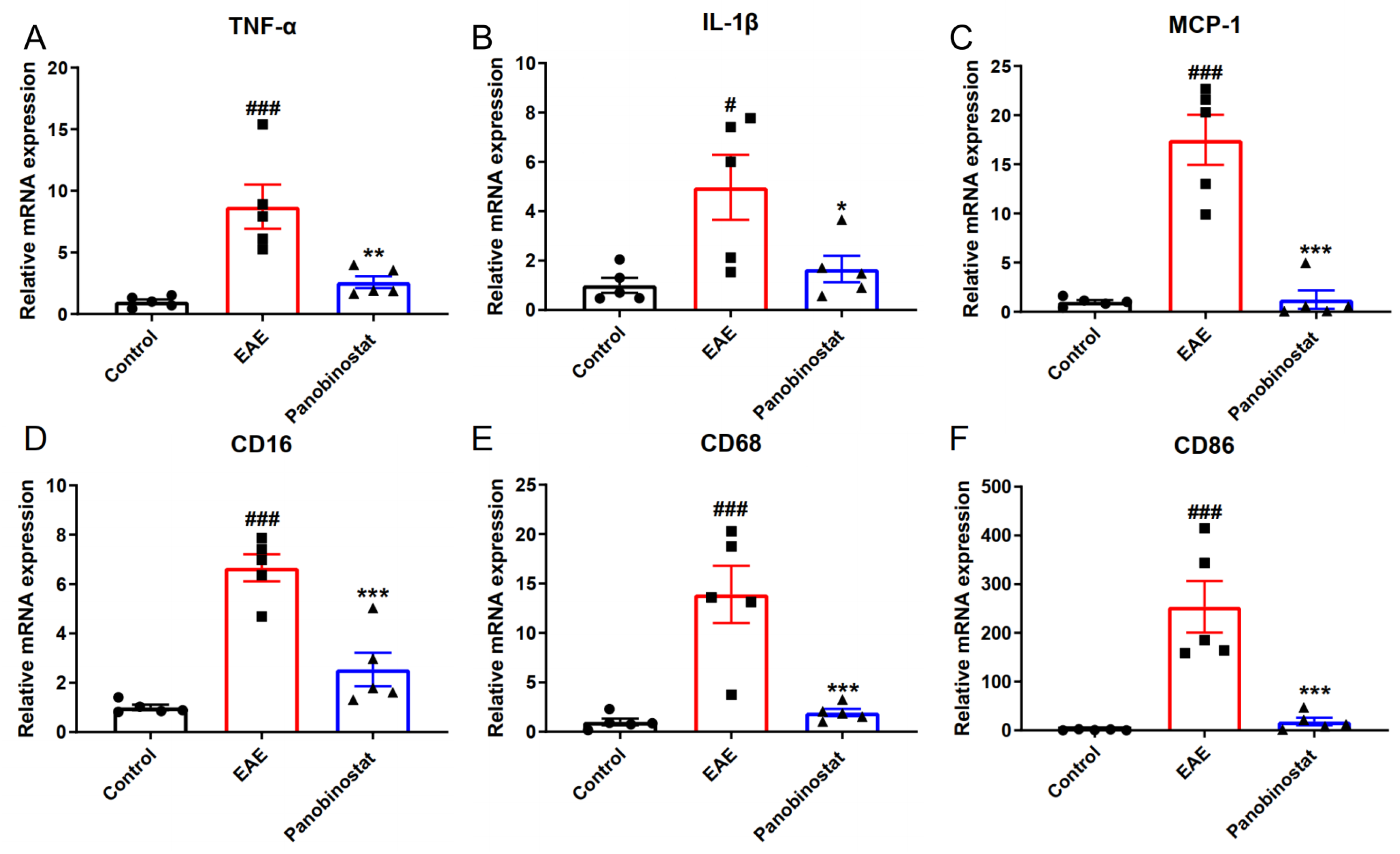
2.4. Panobinostat Reduced the mRNA Level of Pro-Inflammatory Cytokines and Suppressed M1 Microglia Polarization in the Spinal Cord Tissues of EAE Mice
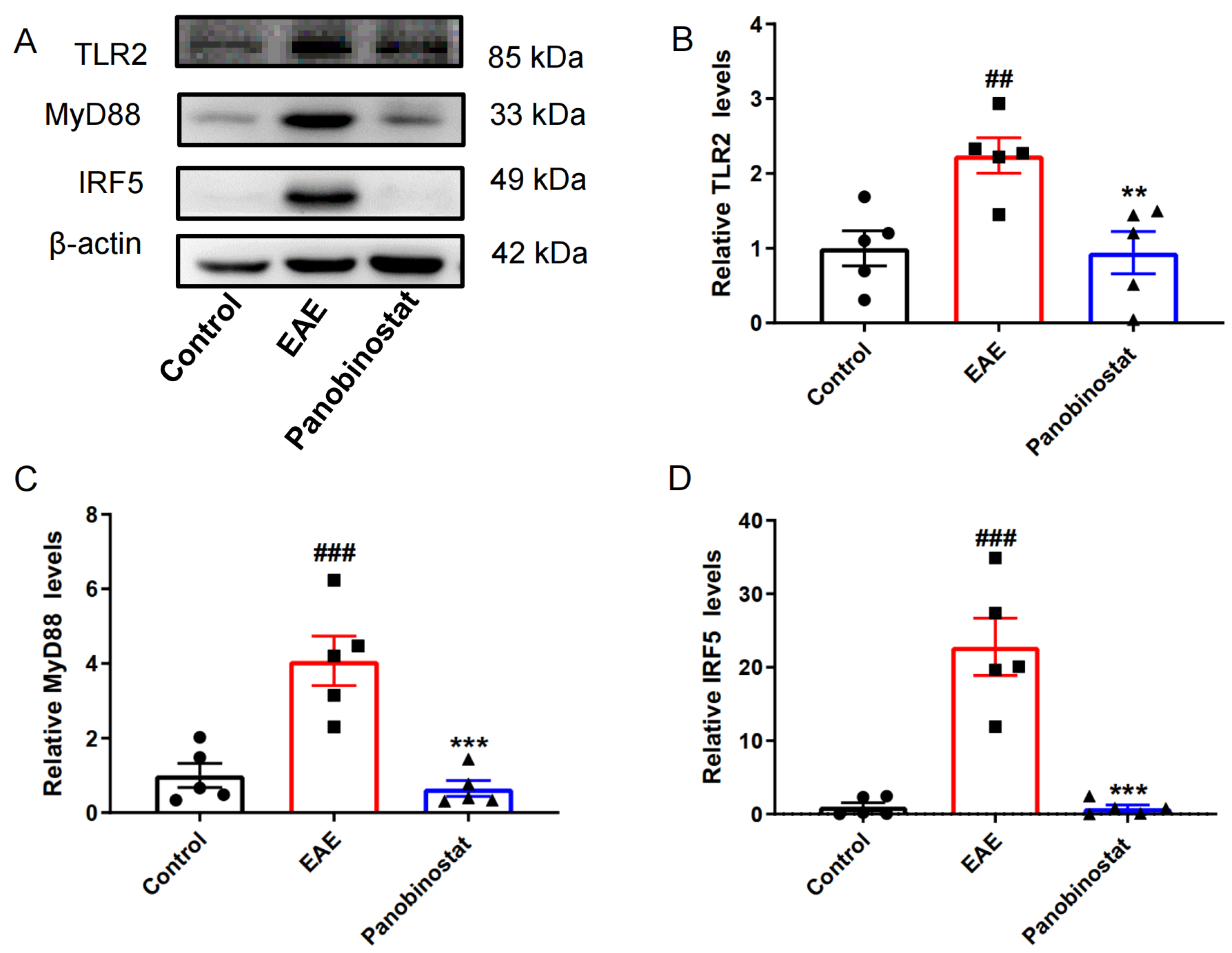
2.5. Panobinostat Inhibited Neuroinflammation via TLR2/MyD88/IRF5 Signaling in the Spinal Cords of EAE Mice
2.6. Panobinostat Mitigated the Impairment of Mitochondrial Function Within the Spinal Cord Tissues of Mice Affected by EAE
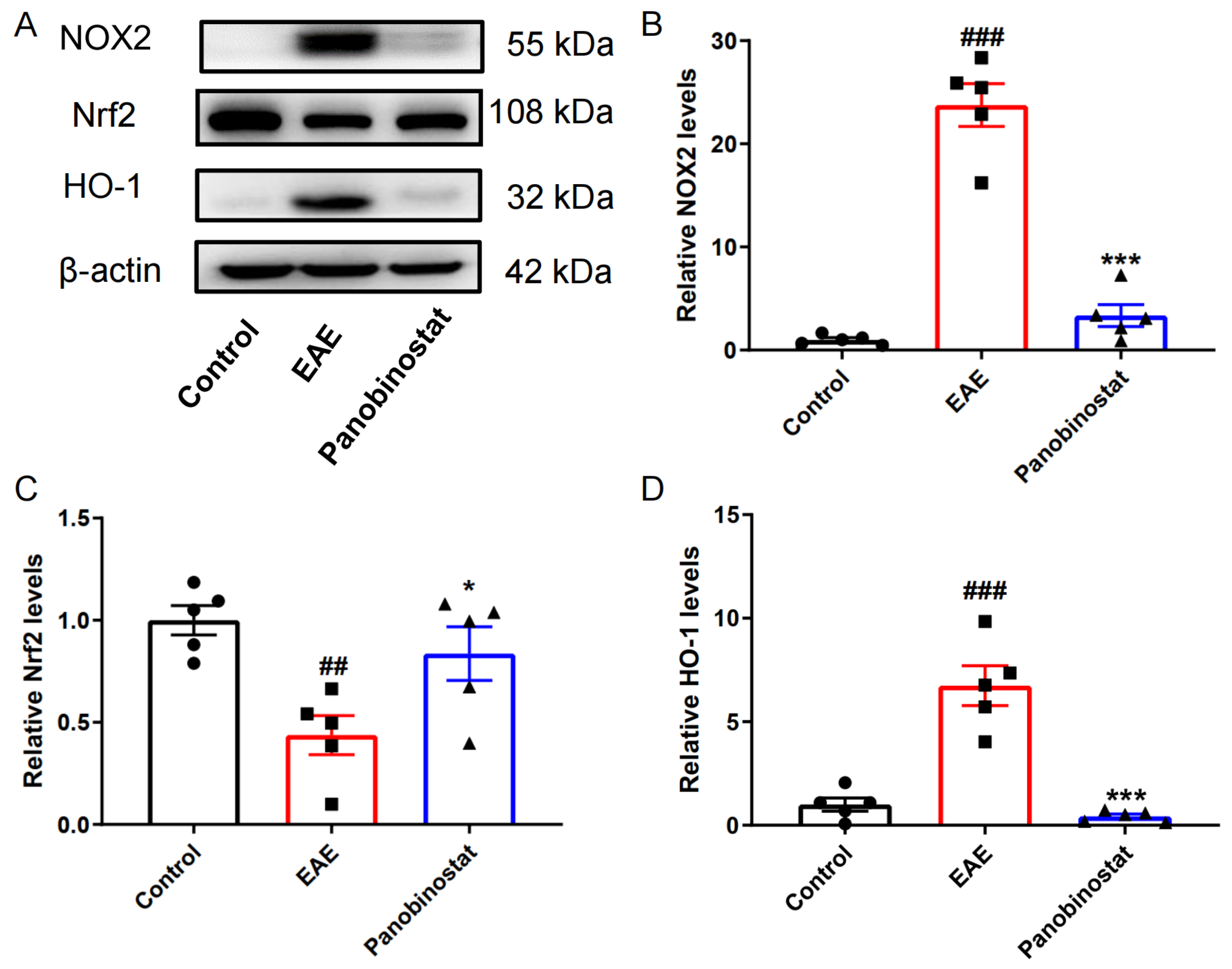
2.7. Panobinostat Mitigated Oxidative Stress Within the Spinal Cord Tissues of Mice with EAE
3. Discussion
4. Materials and Methods
4.1. Animal and Drug
4.2. Induction of EAE Model
4.3. Panobinostat Administration and Groups
4.4. Behavioral Evaluation
4.5. Histology and Immunohistochemistry
4.6. Quantitative Real-Time RT-PCR
4.7. Western Blotting (WB) Analysis
4.8. Statistical Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Haki, M.; Al-Biati, H.A.; Al-Tameemi, Z.S.; Ali, I.S.; Al-Hussaniy, H.A. Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Medicine 2024, 103, e37297. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; Cohen, J.A.; Correale, J.; Graves, J.; Marrie, R.A.; Montalban, X.; Yong, V.W.; Thompson, A.J.; et al. Multiple sclerosis progression: Time for a new mechanism-driven framework. Lancet Neurol. 2023, 22, 78–88. [Google Scholar] [CrossRef]
- Thompson, K.K.; Tsirka, S.E. The Diverse Roles of Microglia in the Neurodegenerative Aspects of Central Nervous System (CNS) Autoimmunity. Int. J. Mol. Sci. 2017, 18, 504. [Google Scholar] [CrossRef]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef]
- Matthews, P.M.; Roncaroli, F.; Waldman, A.; Sormani, M.P.; De Stefano, N.; Giovannoni, G.; Reynolds, R. A practical review of the neuropathology and neuroimaging of multiple sclerosis. Pract. Neurol. 2016, 16, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.S.; Engler, J.B.; Friese, M.A. The neuropathobiology of multiple sclerosis. Nature reviews. Neuroscience 2024, 25, 493–513. [Google Scholar]
- Sun, R.; Wang, Y.F.; Yang, X. Knockdown of IFIT3 ameliorates multiple sclerosis via selectively regulating M1 polarization of microglia in an experimental autoimmune encephalomyelitis model. Int. Immunopharmacol. 2024, 128, 111501. [Google Scholar] [CrossRef]
- Howell, O.W.; Rundle, J.L.; Garg, A.; Komada, M.; Brophy, P.J.; Reynolds, R. Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 1017–1033. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. 2017, 37, 10185–10199. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, K.C.; Osunde, M.; Tiwari-Woodruff, S.K. The complexities of investigating mitochondria dynamics in multiple sclerosis and mouse models of MS. Front. Neurosci. 2023, 17, 1144896. [Google Scholar] [CrossRef] [PubMed]
- Hares, K.; Kemp, K.; Rice, C.; Gray, E.; Scolding, N.; Wilkins, A. Reduced axonal motor protein expression in non-lesional grey matter in multiple sclerosis. Mult. Scler. 2014, 20, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Ramos-González, E.J.; Bitzer-Quintero, O.K.; Ortiz, G.; Hernández-Cruz, J.J.; Ramírez-Jirano, L.J. Relationship between inflammation and oxidative stress and its effect on multiple sclerosis. Neurologia 2024, 39, 292–301. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Tobore, T.O. Oxidative/Nitroxidative Stress and Multiple Sclerosis. J. Mol. Neurosci. 2021, 71, 506–514. [Google Scholar] [CrossRef]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J. Oncol. 2020, 2020, 7131802. [Google Scholar] [CrossRef]
- Brinkmann, C.R.; Højen, J.F.; Rasmussen, T.A.; Kjær, A.S.; Olesen, R.; Denton, P.W.; Østergaard, L.; Ouyang, Z.; Lichterfeld, M.; Yu, X.; et al. Treatment of HIV-Infected Individuals with the Histone Deacetylase Inhibitor Panobinostat Results in Increased Numbers of Regulatory T Cells and Limits Ex Vivo Lipopolysaccharide-Induced Inflammatory Responses. mSphere 2018, 3, 10–128. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Gavathiotis, E. Mitochondrial dynamics proteins as emerging drug targets. Trends Pharmacol. Sci. 2023, 44, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Treuer, A.V.; Faúndez, M.; Ebensperger, R.; Hovelmeyer, E.; Vergara-Jaque, A.; Perera-Sardiña, Y.; Gutierrez, M.; Fuentealba, R.; González, D.R. New NADPH Oxidase 2 Inhibitors Display Potent Activity against Oxidative Stress by Targeting p22(phox)-p47(phox) Interactions. Antioxidants 2023, 12, 1441. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, R.; Zhao, J.; Chen, M.; Chen, S.; Ji, B.; Chen, H.; Liu, D.; Li, L.; Du, G. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/NF-κB p65-mediated neuroinflammation. Pharmacol. Res. 2022, 176, 105969. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Labib, D.A.; Ashmawy, I.; Elmazny, A.; Helmy, H.; Ismail, R.S. Toll-like receptors 2 and 4 expression on peripheral blood lymphocytes and neutrophils of Egyptian multiple sclerosis patients. Int. J. Neurosci. 2022, 132, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Morandi, E.; Tanasescu, R.; Frakich, N.; Caldano, M.; Onion, D.; Faraj, T.A.; Erridge, C.; Gran, B. The Soluble Form of Toll-Like Receptor 2 Is Elevated in Serum of Multiple Sclerosis Patients: A Novel Potential Disease Biomarker. Front. Immunol. 2018, 9, 457. [Google Scholar] [CrossRef]
- Miranda-Hernandez, S.; Gerlach, N.; Fletcher, J.M.; Biros, E.; Mack, M.; Körner, H.; Baxter, A.G. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 791–804. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, J.; Wu, M.; Xu, L.; Wang, Y.; Yuan, W.; Hua, F.; Fan, H.; Dong, F.; Qu, X.; et al. Toll-Like Receptor 4 Promotes Th17 Lymphocyte Infiltration Via CCL25/CCR9 in Pathogenesis of Experimental Autoimmune Encephalomyelitis. J. Neuroimmune Pharmacol. 2019, 14, 493–502. [Google Scholar] [CrossRef]
- Marta, M.; Andersson, Å.; Isaksson, M.; Kämpe, O.; Lobell, A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2008, 38, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain A J. Neurol. 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Recks, M.S.; Stormanns, E.R.; Bader, J.; Arnhold, S.; Addicks, K.; Kuerten, S. Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin. Immunol. 2013, 149, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Grel, H.; Woznica, D.; Ratajczak, K.; Kalwarczyk, E.; Anchimowicz, J.; Switlik, W.; Olejnik, P.; Zielonka, P.; Stobiecka, M.; Jakiela, S. Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes. Int. J. Mol. Sci. 2023, 24, 13033. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Gui, L.N.; Liu, Y.Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxidative Med. Cell. Longev. 2020, 2020, 7191080. [Google Scholar] [CrossRef]
- Kees, F. Dimethyl fumarate: A Janus-faced substance? Expert Opin. Pharmacother. 2013, 14, 1559–1567. [Google Scholar] [CrossRef]
- Ravelli, K.G.; Santos, G.D.; Dos Santos, N.B.; Munhoz, C.D.; Azzi-Nogueira, D.; Campos, A.C.; Pagano, R.L.; Britto, L.R.; Hernandes, M.S. Nox2-dependent Neuroinflammation in An EAE Model of Multiple Sclerosis. Transl. Neurosci. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Buonvicino, D.; Ranieri, G.; Chiarugi, A. Treatment with Non-specific HDAC Inhibitors Administered after Disease Onset does not Delay Evolution in a Mouse Model of Progressive Multiple Sclerosis. Neuroscience 2021, 465, 38–45. [Google Scholar] [CrossRef]
- Aulova, K.S.; Urusov, A.E.; Toporkova, L.B.; Sedykh, S.E.; Shevchenko, Y.A.; Tereshchenko, V.P.; Sennikov, S.V.; Budde, T.; Meuth, S.G.; Popova, N.A.; et al. Production of Abzymes in Th, CBA, and C57BL/6 Mice before and after MOG Treatment: Comparing Changes in Cell Differentiation and Proliferation. Biomolecules 2019, 10, 53. [Google Scholar] [CrossRef]
- Laaker, C.; Hsu, M.; Fabry, Z.; Miller, S.D.; Karpus, W.J. Experimental Autoimmune Encephalomyelitis in the Mouse. Curr. Protoc. 2021, 1, e300. [Google Scholar] [CrossRef] [PubMed]
- Buonvicino, D.; Ranieri, G.; Pratesi, S.; Guasti, D.; Chiarugi, A. Neuroimmunological characterization of a mouse model of primary progressive experimental autoimmune encephalomyelitis and effects of immunosuppressive or neuroprotective strategies on disease evolution. Exp. Neurol. 2019, 322, 113065. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Zhao, J.; Yang, R.; Yang, H.; Guo, M.; Ji, B.; Du, G.; Li, L. Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction. Int. J. Mol. Sci. 2024, 25, 12035. https://doi.org/10.3390/ijms252212035
Shen Y, Zhao J, Yang R, Yang H, Guo M, Ji B, Du G, Li L. Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2024; 25(22):12035. https://doi.org/10.3390/ijms252212035
Chicago/Turabian StyleShen, Yanjia, Jiaying Zhao, Ran Yang, Huilin Yang, Minmin Guo, Baixi Ji, Guanhua Du, and Li Li. 2024. "Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction" International Journal of Molecular Sciences 25, no. 22: 12035. https://doi.org/10.3390/ijms252212035
APA StyleShen, Y., Zhao, J., Yang, R., Yang, H., Guo, M., Ji, B., Du, G., & Li, L. (2024). Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction. International Journal of Molecular Sciences, 25(22), 12035. https://doi.org/10.3390/ijms252212035