
Complete Mitogenomes of Xinjiang Hares and Their Selective Pressure Considerations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
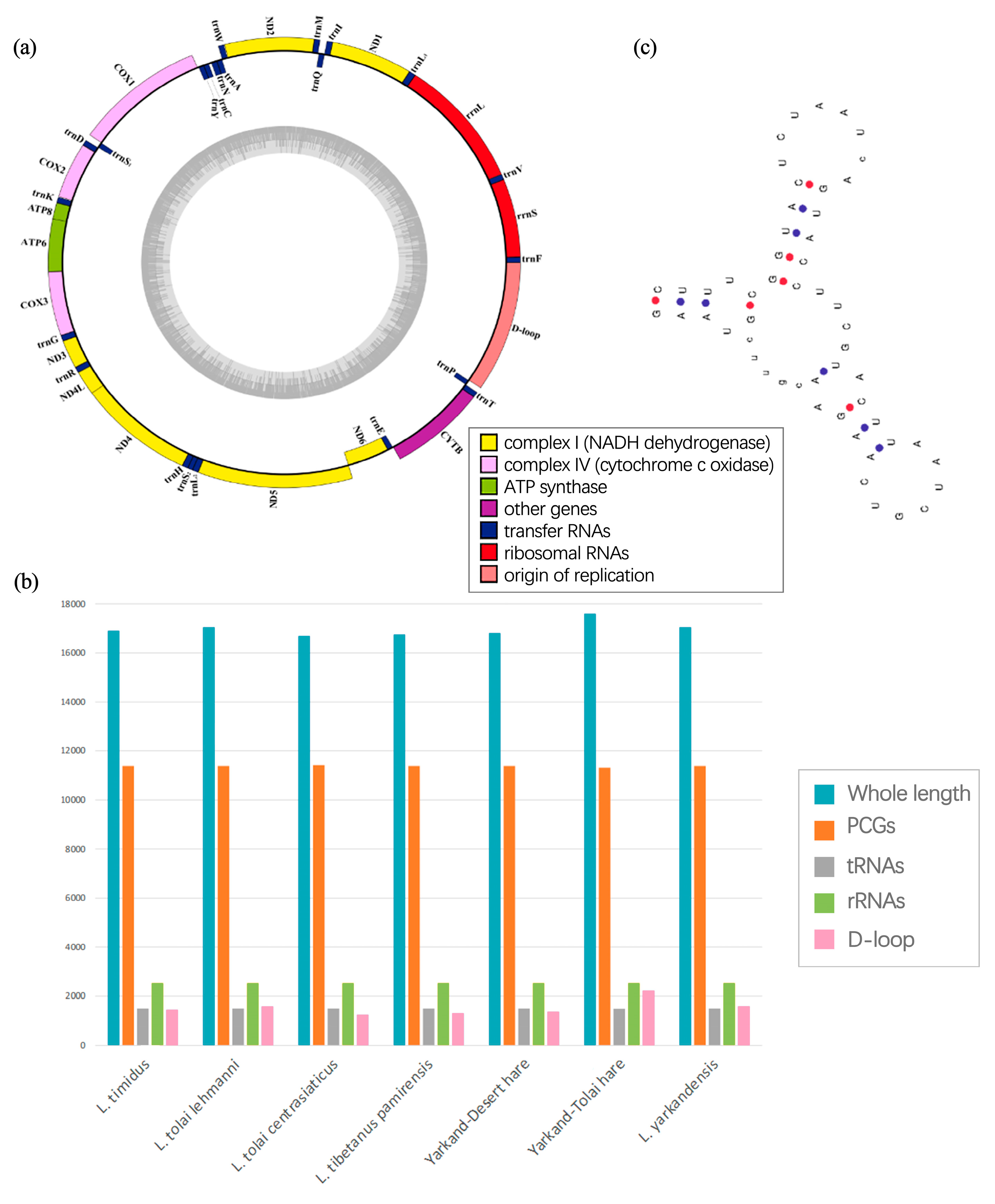
2.1. Mitochondrial Genome Components and Sizes
2.2. tRNA and rRNA
2.3. Base Composition and Skews
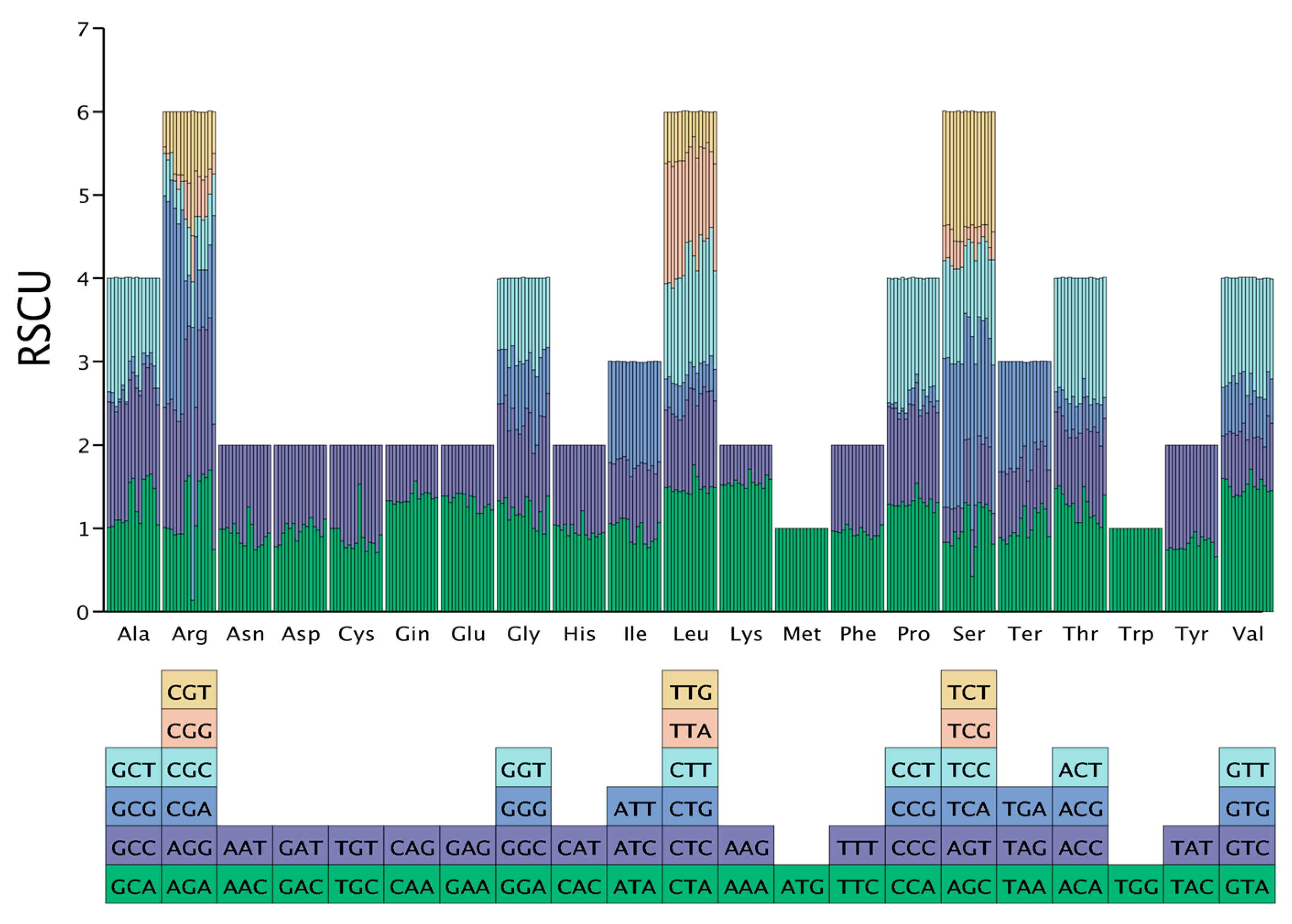
2.4. Relative Synonymous Codon Usage
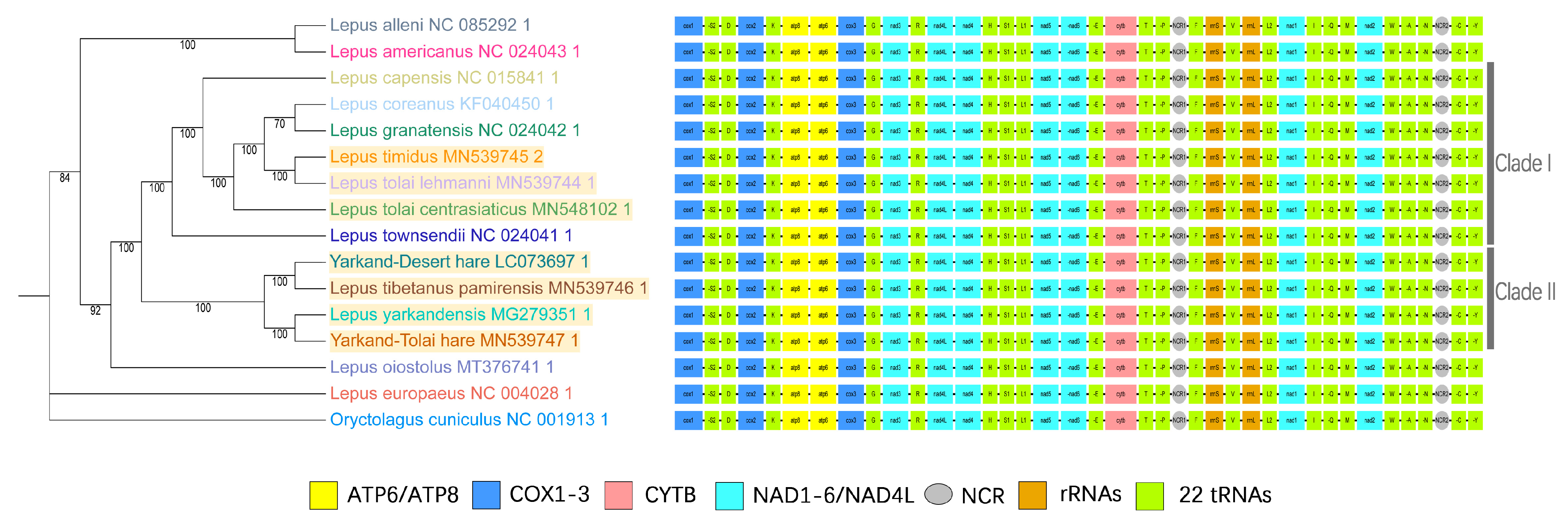
2.5. Phylogenetic Analysis
2.6. Control Region
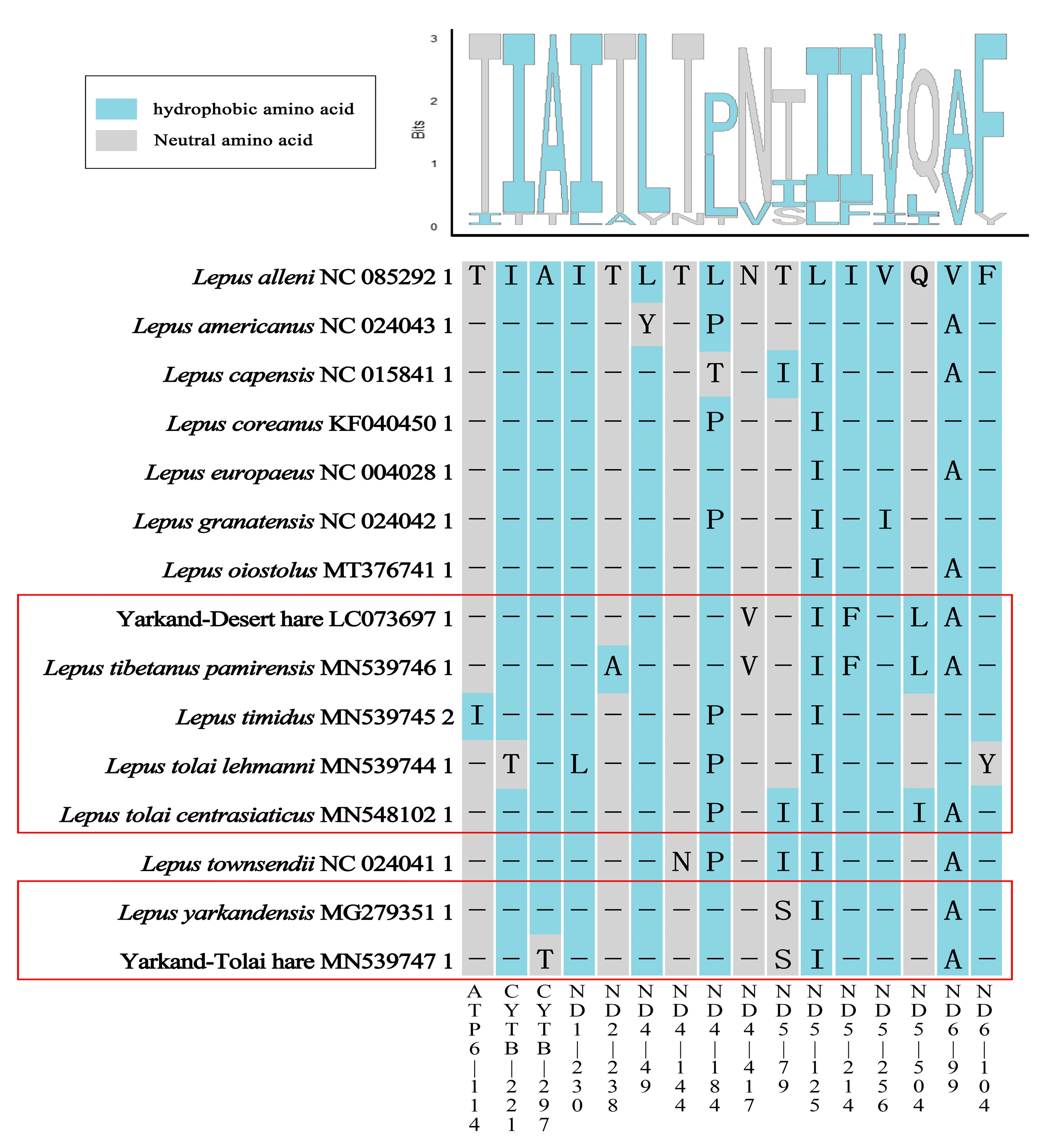
2.7. Selective Pressure on PCGs
3. Discussion
3.1. Hare Mitochondrial Genome Phylogeny Analysis
3.2. Mitochondrial Genome Components
3.3. Adaptive Evolution
4. Materials and Methods
4.1. Sample Collection and Sequencing Assembly
4.2. Mitochondrial Genome Structure Analysis
4.3. Comparative Analysis of the Mitochondrial Genome of Lepus Species
4.4. Phylogenetic Analysis
4.5. Mitochondrial Genome Control Region
4.6. Selective Pressure on PCGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, J.; Chen, Y.; Guan, X.; Zhao, Y.; Chen, J.; Mao, W. Recent climate and hydrological changes in a mountain–basin system in Xinjiang, China. Earth-Sci. Rev. 2022, 226, 103957. [Google Scholar] [CrossRef]
- Guan, J.; Yao, J.; Li, M.; Li, D.; Zheng, J. Historical changes and projected trends of extreme climate events in Xinjiang, China. Clim. Dyn. 2022, 59, 1753–1774. [Google Scholar] [CrossRef]
- Shan, W.J.; Li, Z.R.; Dai, H.Y.; Dong, P.C.; Zhang, Y.C. Taxonomic status of hares (Lepus spp.) in Xinjiang, China (Lagomorpha: Leporidae): An integrative approach. Zool. Scr. 2024, 53, 282–298. [Google Scholar] [CrossRef]
- Smith, A.; Johnston, C.; Alves, P.; Hackländer, K. Lagomorphs: Pikas, Rabbits, and Hares of the World; JHU Press: Baltimore, MD, USA, 2018; pp. 1–266. [Google Scholar]
- Mamat, M.; Shan, W.; Dong, P.; Zhou, S.; Liu, P.; Meng, Y.; Nie, W.; Teng, P.; Zhang, Y. Population genetics analysis of Tolai hares (Lepus tolai) in Xinjiang, China using genome-wide SNPs from SLAF-seq and mitochondrial markers. Front. Genet. 2022, 13, 1018632. [Google Scholar]
- Li, Z.; Fang, B.; Dong, P.; Shan, W. Selective sweep analysis of the adaptability of the Yarkand hare (Lepus yarkandensis) to hot arid environments using SLAF-seq. Anim. Genet. 2024, 55, 681–686. [Google Scholar] [CrossRef]
- Ertl, H. Nuclear genome influences mitochondrial DNA. Nat. Rev. Genet. 2023, 24, 803. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, W.; Zhang, Y.; Wang, A.; Liu, C.; Gu, Z.; Yang, Y. The molecular phylogeny of Caenogastropoda (Mollusca, Gastropoda) based on mitochondrial genomes and nuclear genes. Gene 2024, 928, 148790. [Google Scholar] [CrossRef]
- Zuo, W.; Li, H. Assemble and comparative analysis of the mitochondrial genome of Rhododendron delavayi: Insights into phylogenetic relationships and genomic variations. Gene 2024, 927, 148741. [Google Scholar] [CrossRef]
- Murat, F.; Mbengue, N.; Winge, S.B.; Trefzer, T.; Leushkin, E.; Sepp, M.; Cardoso-Moreira, M.; Schmidt, J.; Schneider, C.; Mößinger, K.; et al. The molecular evolution of spermatogenesis across mammals. Nature 2023, 613, 308–316. [Google Scholar] [CrossRef]
- Granath-Panelo, M.; Kajimura, S. Mitochondrial heterogeneity and adaptations to cellular needs. Nat. Cell Biol. 2024, 26, 674–686. [Google Scholar] [CrossRef]
- Goh, K.M.; González-Siso, M.-I.; Sani, R.K. Genomics of extreme environments: Unveiling the secrets of survival. Sci. Rep. 2023, 13, 21441. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Tursun, M.; Zhou, S.; Zhang, Y.; Dai, H. The complete mitochondrial genome sequence of Lepus tolai in Xinjiang. Mitochondrial DNA Part B 2020, 5, 1336–1337. [Google Scholar] [CrossRef]
- Shan, W.; Tursun, M.; Zhou, S.; Zhang, Y.; Dai, H. Complete mitochondrial genome sequence of Lepus yarkandensis Günther, 1875 (Lagomorpha, Leporidae): Characterization and phylogenetic analysis. Zookeys 2021, 1012, 135–150. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, L.; Zhang, Q.; Yang, Q.; Meng, X. Bidirectional introgressive hybridization between Lepus capensis and Lepus yarkandensis. Mol. Phylogenetics Evol. 2011, 59, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Melo-Ferreira, J.; Boursot, P.; Carneiro, M.; Esteves, P.J.; Farelo, L.; Alves, P.C. Recurrent introgression of mitochondrial DNA among hares (Lepus spp.) revealed by species-tree inference and coalescent simulations. Syst. Biol. 2012, 61, 367–381. [Google Scholar] [CrossRef]
- Huang, Y.L.; Chen, Y.X.; Guo, H.T.; Xu, Y.H.; Liu, H.Y.; Liu, D.W. The complete mitochondrial genome sequence of Yarkand hare (Lepus yarkandensis). Mitochondrial DNA B Resour. 2019, 4, 3727–3728. [Google Scholar] [CrossRef]
- Yu, J.N.; Chung, C.U.; Kwak, M. The complete mitochondrial genome sequence of the Korean hare (Lepus coreanus). Mitochondrial DNA 2015, 26, 129–130. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, L.; Guo, S. The sequence and characterization of mitochondrial of Lepus oiostolus (Lagomorpha: Leporidae). Mitochondrial DNA B Resour. 2020, 5, 2135–2136. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Chen, M.; Pan, T.; Zhang, B.; Zhou, Y.; Wang, H. Complete mitochondrial DNA sequence of Lepus sinensis (Leporidae: Lepus). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 1711–1712. [Google Scholar] [CrossRef]
- Kinoshita, G.; Nunome, M.; Kryukov, A.P.; Kartavtseva, I.V.; Han, S.-H.; Yamada, F.; Suzuki, H. Contrasting phylogeographic histories between the continent and islands of East Asia: Massive mitochondrial introgression and long-term isolation of hares (Lagomorpha: Lepus). Mol. Phylogenetics Evol. 2019, 136, 65–75. [Google Scholar] [CrossRef]
- Cheng, C.; Ge, D.; Xia, L.; Zhou, C.; Yang, Q. Morphometrics study on the so called ‘Cape hare’ (Lagomorpha: Leporidae: Lepus) in China. Acta Theriol. Sin. 2012, 32, 275–286. [Google Scholar]
- Shan, W.J.; Liu, J.; Halik, M. Genetic structure and subspecies divergence of Lepus capensis in Xinjiang. Dongwuxue Yanjiu 2011, 32, 179–187. [Google Scholar] [PubMed]
- Zhu, Y.; Yan, S.; Ma, P.; Zhang, Y.; Zuo, C.; Ma, X.; Zhang, Z. Comparative mitochondrial genome analysis provides new insights into the classification of Modiolinae. Mol. Biol. Rep. 2024, 51, 823. [Google Scholar] [CrossRef]
- Cui, P.; Ji, R.; Ding, F.; Qi, D.; Gao, H.; Meng, H.; Yu, J.; Hu, S.; Zhang, H. A complete mitochondrial genome sequence of the wild two-humped camel (Camelus bactrianus ferus): An evolutionary history of camelidae. BMC Genom. 2007, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Zeng, B.; Meng, X.; Yue, B.; Zhang, Z.; Zou, F. The complete mitochondrial genome and phylogenetic analysis of the giant panda (Ailuropoda melanoleuca). Gene 2007, 397, 76–83. [Google Scholar] [CrossRef]
- Hu, X.D.; Gao, L.Z. The complete mitochondrial genome of domestic sheep, Ovis aries. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 1425–1427. [Google Scholar] [CrossRef]
- Xiao, X.; Yang, S. The Complete Mitochondrial Genome and Phylogenetic Analysis of Chinese Jianchang Horse (Equus caballus). Cloning Transgenesis 2015, 5, 149. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, H.; Mayden, R.; Xiong, B. Comparison of evolutionary rates in the mitochondrial DNA cytochrome b gene and control region and their implications for phylogeny of the Cobitoidea (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 2006, 39, 347–357. [Google Scholar] [CrossRef]
- Iyengar, A.; Diniz, F.M.; Gilbert, T.; Woodfine, T.; Knowles, J.; Maclean, N. Structure and evolution of the mitochondrial control region in oryx. Mol. Phylogenetics Evol. 2006, 40, 305–314. [Google Scholar] [CrossRef]
- Sbisà, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Sun, K.; Feng, J.; Jin, L.; Liu, Y.; Shi, L.; Jiang, T. Structure, DNA sequence variation and phylogenetic implications of the mitochondrial control region in horseshoe bats. Mamm. Biol. 2009, 74, 130–144. [Google Scholar] [CrossRef]
- Zhang, H.H.; Xu, C.; Ma, J.Z. Structure of the mtDNA control region and phylogeny of the Mustelidae species. Acta Ecol. Sin. 2009, 29, 3585–3592. [Google Scholar]
- Liu, Y.; Yang, Q.; Zhao, F. Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 2021, 90, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Panja, A.S.; Bandopadhyay, B.; Maiti, S. Protein Thermostability Is Owing to Their Preferences to Non-Polar Smaller Volume Amino Acids, Variations in Residual Physico-Chemical Properties and More Salt-Bridges. PLoS ONE 2015, 10, e0131495. [Google Scholar] [CrossRef]
- Tyagi, A.; Ali, S.; Ramakrishna, G.; Singh, A.; Park, S.; Mahmoudi, H.; Bae, H. Revisiting the Role of Polyamines in Plant Growth and Abiotic Stress Resilience: Mechanisms, Crosstalk, and Future Perspectives. J. Plant Growth Regul. 2022, 42, 5074–5098. [Google Scholar] [CrossRef]
- Shim, J.S.; Jeong, H.I.; Bang, S.W.; Jung, S.E.; Kim, G.; Kim, Y.S.; Redillas, M.; Oh, S.J.; Seo, J.S.; Kim, J.K. Drought-Induced Branched-Chain Amino Acid Aminotransferase enhances drought tolerance in rice. Plant Physiol. 2023, 191, 1435–1447. [Google Scholar] [CrossRef]
- Catalano, C.; Al Mughram, M.H.; Guo, Y.; Kellogg, G.E. 3D interaction homology: Hydropathic interaction environments of serine and cysteine are strikingly different and their roles adapt in membrane proteins. Curr. Res. Struct. Biol. 2021, 3, 239–256. [Google Scholar] [CrossRef]
- Dinkeloo, K.; Boyd, S.; Pilot, G. Update on amino acid transporter functions and on possible amino acid sensing mechanisms in plants. Semin. Cell Dev. Biol. 2018, 74, 105–113. [Google Scholar] [CrossRef]
- Senkler, J.; Senkler, M.; Braun, H.P. Structure and function of complex I in animals and plants—A comparative view. Physiol. Plant 2017, 161, 6–15. [Google Scholar] [CrossRef]
- Formosa, L.E.; Ryan, M.T. Mitochondrial OXPHOS complex assembly lines. Nat. Cell Biol. 2018, 20, 511–513. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rodriguez, M.; Martinez, C.A.; Roca, J.; Rodriguez-Martinez, H. mRNA expression of oxidative-reductive proteins in boars with documented different fertility can identify relevant prognostic biomarkers. Res. Vet. Sci. 2021, 141, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F.; Kirnos, M.D. Structure of animal mitochondrial DNA (base composition, pyrimidine clusters, character of methylation). Biochim. Biophys. Acta 1977, 475, 323–336. [Google Scholar] [CrossRef]
- Bahbahani, H.; Al-Zoubi, S.; Ali, F.; Afana, A.; Dashti, M.; Al-Ateeqi, A.; Wragg, D.; Al-Bustan, S.; Almathen, F. Signatures of purifying selection and site-specific positive selection on the mitochondrial DNA of dromedary camels (Camelus dromedarius). Mitochondrion 2023, 69, 36–42. [Google Scholar] [CrossRef]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Rould, M.A.; Perona, J.J.; Steitz, T.A. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature 1991, 352, 213–218. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Sau, K. CodonU: A Python Package for Codon Usage Analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2024, 21, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Wu, J.; Lu, M.; Tian, T.; Wang, D.; Luo, Z.; Yang, D.; Li, L.; Yang, X.; Liu, D.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Fritillaria ussuriensis Maxim. (Liliales: Liliaceae), an endangered medicinal plant. BMC Genom. 2024, 25, 773. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Marshall, H.D.; Baker, A.J. Structural conservation and variation in the mitochondrial control region of fringilline finches (Fringilla spp.) and the greenfinch (Carduelis chloris). Mol. Biol. Evol. 1997, 14, 173–184. [Google Scholar] [CrossRef]
- Tan, B.G.; Gustafsson, C.M.; Falkenberg, M. Mechanisms and regulation of human mitochondrial transcription. Nat. Rev. Mol. Cell Biol. 2024, 25, 119–132. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Tursun, M.; Shan, W. Complete Mitogenomes of Xinjiang Hares and Their Selective Pressure Considerations. Int. J. Mol. Sci. 2024, 25, 11925. https://doi.org/10.3390/ijms252211925
Wang R, Tursun M, Shan W. Complete Mitogenomes of Xinjiang Hares and Their Selective Pressure Considerations. International Journal of Molecular Sciences. 2024; 25(22):11925. https://doi.org/10.3390/ijms252211925
Chicago/Turabian StyleWang, Ruijie, Mayinur Tursun, and Wenjuan Shan. 2024. "Complete Mitogenomes of Xinjiang Hares and Their Selective Pressure Considerations" International Journal of Molecular Sciences 25, no. 22: 11925. https://doi.org/10.3390/ijms252211925
APA StyleWang, R., Tursun, M., & Shan, W. (2024). Complete Mitogenomes of Xinjiang Hares and Their Selective Pressure Considerations. International Journal of Molecular Sciences, 25(22), 11925. https://doi.org/10.3390/ijms252211925