
Wakefulness Induced by TAAR1 Partial Agonism in Mice Is Mediated Through Dopaminergic Neurotransmission †


Abstract
1. Introduction
2. Results
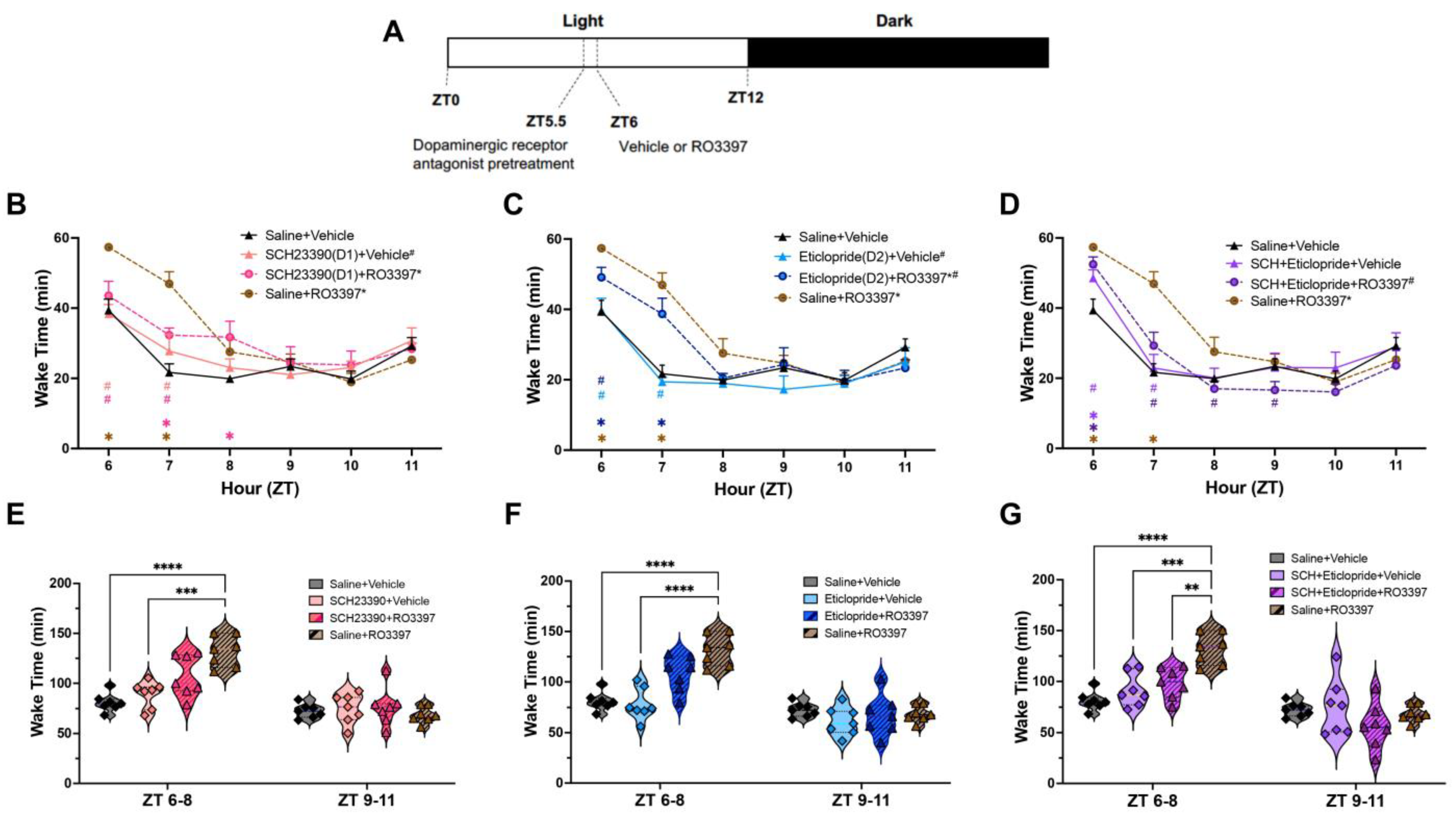
2.1. Wakefulness
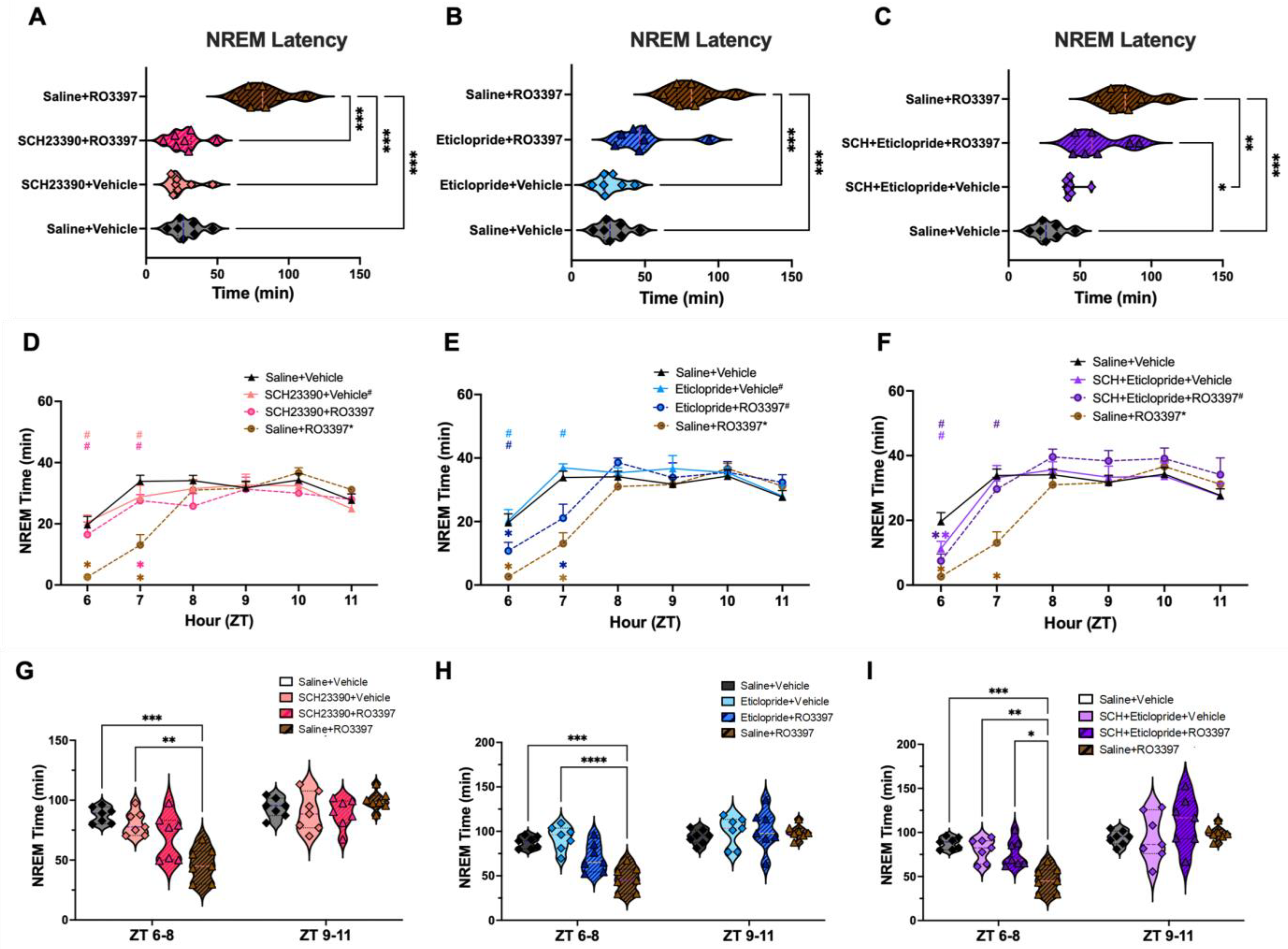
2.2. NREM Sleep
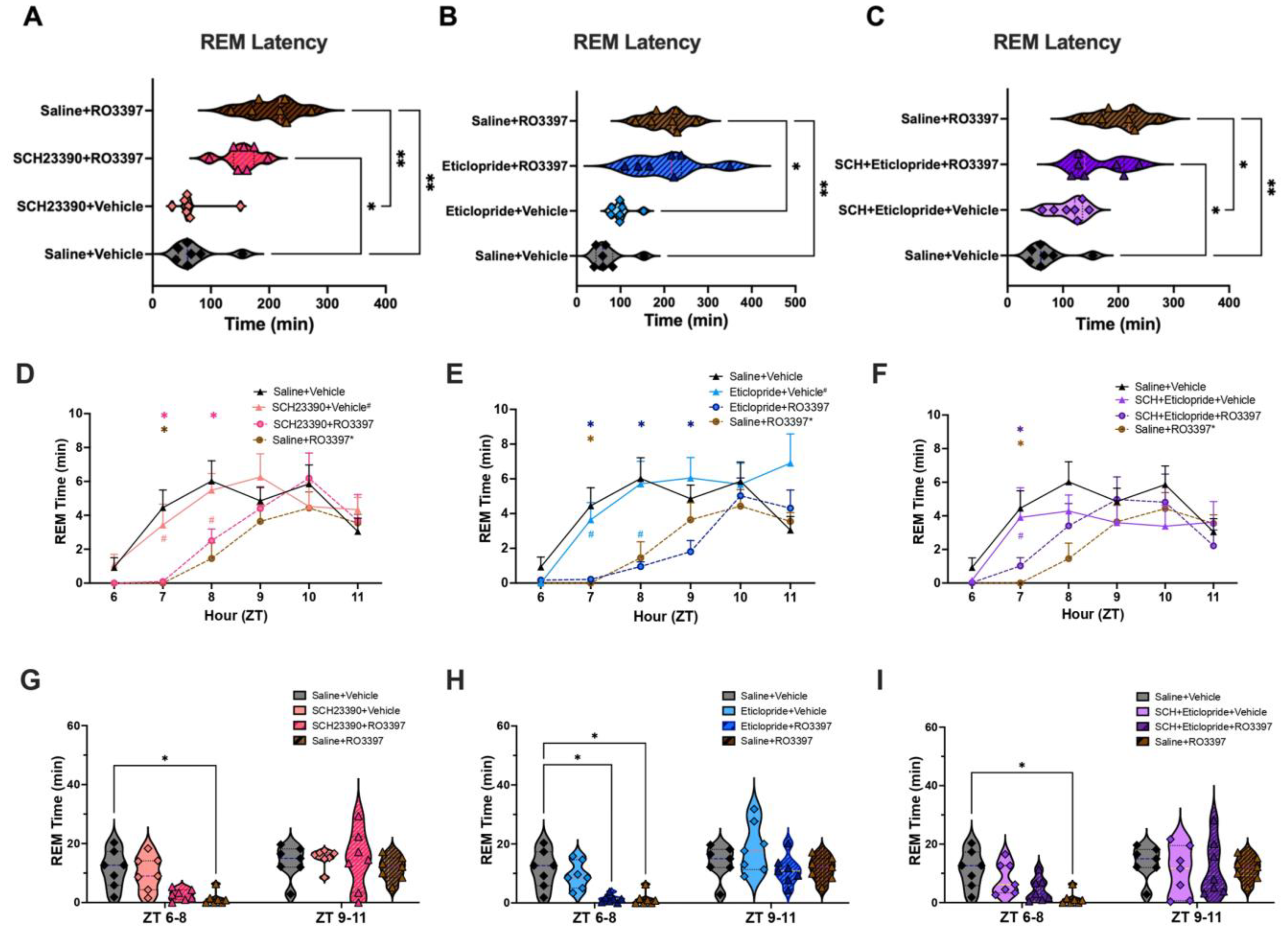
2.3. REM Sleep
2.4. EEG Spectral Analysis
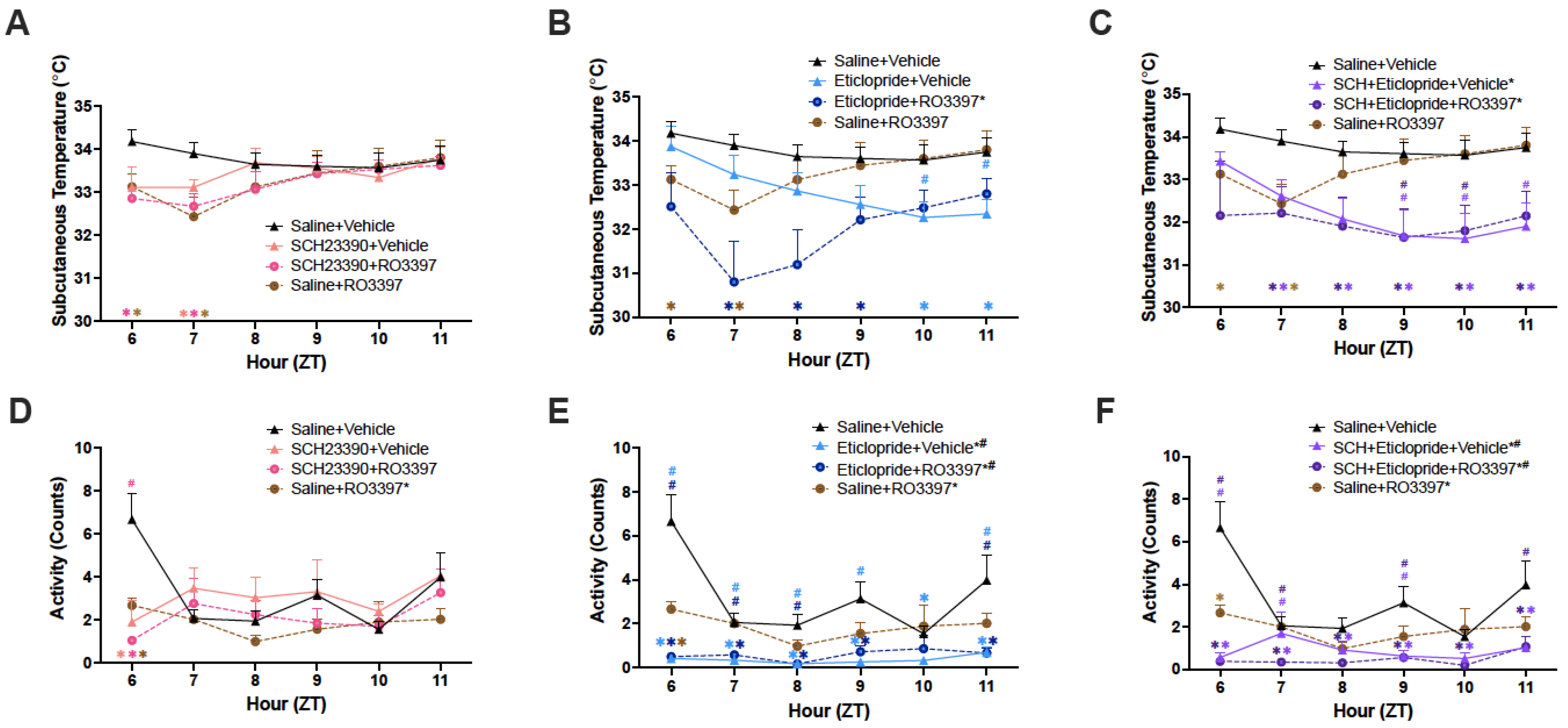
2.5. Subcutaneous Temperature (Tsc)
2.6. Activity
3. Discussion
3.1. Dopaminergic Effects on Sleep/Wake Control in Rodents
3.2. Dopaminergic Antagonism Reduces TAAR1-Mediated Wakefulness
3.3. Distinct Dopaminergic Effects on TAAR1-Mediated NREM Sleep Latency vs. NREM Sleep Time
3.4. Absence of Dopaminergic Involvement in TAAR1 Effects on REM Sleep or EEG Spectra
3.5. Tsc and Activity
3.6. Differential Effects of D1 vs. D2 Antagonism on TAAR1-Mediated Effects
3.7. Limitations of the Present Study
4. Materials and Methods
4.1. Animals
4.2. Surgical Procedures
4.3. EEG, EMG, LMA and Tsc Recording
4.4. Experimental Protocols
4.5. Drugs
4.6. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, M.D. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 2004, 90, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef]
- Lindemann, L.; Ebeling, M.; Kratochwil, N.A.; Bunzow, J.R.; Grandy, D.K.; Hoener, M.C. Trace amine-associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics 2005, 85, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Liberles, S.D.; Buck, L.B. A second class of chemosensory receptors in the olfactory epithelium. Nature 2006, 442, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.L.; et al. Trace amine-associated receptor 1 modulates dopaminergic activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef]
- Espinoza, S.; Lignani, G.; Caffino, L.; Maggi, S.; Sukhanov, I.; Leo, D.; Mus, L.; Emanuele, M.; Ronzitti, G.; Harmeier, A.; et al. TAAR1 Modulates Cortical Glutamate NMDA Receptor Function. Neuropsychopharmacology 2015, 40, 2217–2227. [Google Scholar] [CrossRef]
- Achat-Mendes, C.; Lynch, L.J.; Sullivan, K.A.; Vallender, E.J.; Miller, G.M. Augmentation of methamphetamine-induced behaviors in transgenic mice lacking the trace amine-associated receptor 1. Pharmacol. Biochem. Behav. 2012, 101, 201–207. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Zorina, O.I.; Ghisi, V.; Caron, M.G.; Gainetdinov, R.R. Trace amine associated receptor 1 and movement control. Park. Relat. Disord. 2008, 14 (Suppl. 2), S99–S102. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, T.D.; Swanson, C.J.; Smith, K.E.; Zhong, H.; Borowsky, B.; Seeman, P.; Branchek, T.; Gerald, C.P. The Trace Amine 1 receptor knockout mouse: An animal model with relevance to schizophrenia. Genes. Brain Behav. 2007, 6, 628–639. [Google Scholar] [CrossRef] [PubMed]
- di Cara, B.; Maggio, R.; Aloisi, G.; Rivet, J.M.; Lundius, E.G.; Yoshitake, T.; Svenningsson, P.; Brocco, M.; Gobert, A.; de Groote, L.; et al. Genetic Deletion of Trace Amine 1 Receptors Reveals Their Role in Auto-Inhibiting the Actions of Ecstasy (MDMA). J. Neurosci. 2011, 31, 16928–16940. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, I.; Dorofeikova, M.; Dolgorukova, A.; Dorotenko, A.; Gainetdinov, R.R. Trace Amine-Associated Receptor 1 Modulates the Locomotor and Sensitization Effects of Nicotine. Front. Pharmacol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Sukhanov, I.; Dorotenko, A.; Dolgorukova, A.; Dorofeikova, M.; Gainetdinov, R.R. The trace amine-associated receptor 1 modulates nicotine behavioural effects. Eur. Neuropsychopharmacol. 2017, 27, S673–S674. [Google Scholar] [CrossRef]
- Revel, F.G.; Meyer, C.A.; Bradaia, A.; Jeanneau, K.; Calcagno, E.; André, C.B.; Haenggi, M.; Miss, M.-T.; Galley, G.; Norcross, R.D.; et al. Brain-Specific Overexpression of Trace Amine-Associated Receptor 1 Alters Monoaminergic Neurotransmission and Decreases Sensitivity to Amphetamine. Neuropsychopharmacology 2012, 37, 2580–2592. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Ferragud, A.; Velázquez-Sánchez, C.; Sotnikova, T.D.; Morairty, S.R.; Harmeier, A.; Groebke Zbinden, K.; Norcross, R.D.; et al. Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics. Biol. Psychiatry 2012, 72, 934–942. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.L.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Groebke Zbinden, K.; Galley, G.; et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2013, 18, 543–556. [Google Scholar] [CrossRef]
- Feemster, J.C.; Westerland, S.M.; Gossard, T.R.; Steele, T.A.; Timm, P.C.; Jagielski, J.T.; Strainis, E.; McCarter, S.J.; Hopkins, S.C.; Koblan, K.S.; et al. Treatment with the novel TAAR1 agonist ulotaront is associated with reductions in quantitative polysomnographic REM sleep without atonia in healthy human subjects: Results of a post-hoc analysis. Sleep Med. 2023, 101, 578–586. [Google Scholar] [CrossRef]
- Kane, J.M. A New Treatment Paradigm: Targeting Trace Amine-Associated Receptor 1 (TAAR1) in Schizophrenia. J. Clin. Psychopharmacol. 2022, 42 (Suppl. 1), S1–S13. [Google Scholar] [CrossRef]
- Dahan, L.; Astier, B.; Vautrelle, N.; Urbain, N.; Kocsis, B.; Chouvet, G. Prominent Burst Firing of Dopaminergic Neurons in the Ventral Tegmental Area during Paradoxical Sleep. Neuropsychopharmacology 2007, 32, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Eban-Rothschild, A.; Rothschild, G.; Giardino, W.J.; Jones, J.R.; De Lecea, L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat. Neurosci. 2016, 19, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, E.; Miyasaka, A.; Sakurai, K.; Cherasse, Y.; Li, Y.; Sakurai, T. Rapid eye movement sleep is initiated by basolateral amygdala dopamine signaling in mice. Science 2022, 375, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Farber, J.; Gatz, P.; Roffwarg, H.; German, D.C. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat. Brain Res. 1983, 273, 133–141. [Google Scholar] [CrossRef]
- Black, S.W.; Schwartz, M.D.; Chen, T.M.; Hoener, M.C.; Kilduff, T.S. Trace Amine-Associated Receptor 1 Agonists as Narcolepsy Therapeutics. Biol. Psychiatry 2017, 82, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.D.; Black, S.W.; Fisher, S.P.; Palmerston, J.B.; Morairty, S.R.; Hoener, M.C.; Kilduff, T.S. Trace Amine-Associated Receptor 1 Regulates Wakefulness and EEG Spectral Composition. Neuropsychopharmacology 2017, 42, 1305–1314. [Google Scholar] [CrossRef]
- Goonawardena, A.V.; Morairty, S.R.; Dell, R.; Orellana, G.A.; Hoener, M.C.; Wallace, T.L.; Kilduff, T.S. Trace amine-associated receptor 1 agonism promotes wakefulness without impairment of cognition in Cynomolgus macaques. Neuropsychopharmacology 2019, 44, 1485–1493. [Google Scholar] [CrossRef]
- Trampus, M.; Ongini, E. The D1 dopamine receptor antagonist SCH 23390 enhances REM sleep in the rat. Neuropharmacology 1990, 29, 889–893. [Google Scholar] [CrossRef]
- Bo, P.; Ongini, E.; Giorgetti, A.; Savoldi, F. Synchronization of the EEG and sedation induced by neuroleptics depend upon blockade of both D-1 and D-2 dopamine receptors. Neuropharmacology 1988, 27, 799–805. [Google Scholar] [CrossRef]
- Eder, D.N.; Zdravkovic, M.; Wildschiødtz, G. Selective alterations of the first NREM sleep cycle in humans by a dopamine D1 receptor antagonist (NNC-687). J. Psychiatric. Res. 2003, 37, 305–312. [Google Scholar] [CrossRef]
- Ongini, E.; Bonizzoni, E.; Ferri, N.; Milani, S.; Trampus, M. Differential effects of dopamine D-1 and D-2 receptor antagonist antipsychotics on sleep-wake patterns in the rat. J. Pharmacol. Exp. Ther. 1993, 266, 726–731. [Google Scholar] [PubMed]
- Bagetta, G.; Corasaniti, M.T.; Strongoli, M.C.; Sakurada, S.; Nisticò, G. Behavioural and ECoG spectrum power effects after intraventricular injection of drugs altering dopaminergic transmission in rats. Neuropharmacology 1987, 26, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Ongini, E.; Bo, P.; Dionisotti, S.; Trampus, M.; Savoldi, F. Effects of remoxipride, a dopamine D-2 antagonist antipsychotic, on sleep-waking patterns and EEG activity in rats and rabbits. Psychopharmacology 1992, 107, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Burgess, C.R.; Tse, G.; Gillis, L.; Peever, J.H. Dopaminergic regulation of sleep and cataplexy in a murine model of narcolepsy. Sleep 2010, 33, 1295–1304. [Google Scholar] [CrossRef]
- Sun, Y.; Tisdale, R.K.; Yamashita, A.; Kilduff, T.S. Peripheral vs. Core Body Temperature as Hypocretin/Orexin Neurons Degenerate: Exercise Mitigates Increased Heat Loss. Peptides 2023, 164, 171002. [Google Scholar] [CrossRef]
- Asif-Malik, A.; Hoener, M.C.; Canales, J.J. Interaction Between the Trace Amine-Associated Receptor 1 and the Dopamine D(2) Receptor Controls Cocaine’s Neurochemical Actions. Sci. Rep. 2017, 7, 13901. [Google Scholar] [CrossRef]
- Bradaia, A.; Trube, G.; Stalder, H.; Norcross, R.D.; Ozmen, L.; Wettstein, J.G.; Pinard, A.; Buchy, D.; Gassmann, M.; Hoener, M.C.; et al. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. USA 2009, 106, 20081–20086. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, S.; Ghisi, V.; Emanuele, M.; Leo, D.; Sukhanov, I.; Sotnikova, T.D.; Chieregatti, E.; Gainetdinov, R.R. Postsynaptic D2 dopamine receptor supersensitivity in the striatum of mice lacking TAAR1. Neuropharmacology 2015, 93, 308–313. [Google Scholar] [CrossRef]
- Espinoza, S.; Salahpour, A.; Masri, B.; Sotnikova, T.D.; Messa, M.; Barak, L.S.; Caron, M.G.; Gainetdinov, R.R. Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol. Pharmacol. 2011, 80, 416–425. [Google Scholar] [CrossRef]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.C.; Sotnikova, T.D.; Gainetdinov, R.R. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef]
- Leo, D.; Sukhanov, I.; Zoratto, F.; Illiano, P.; Caffino, L.; Sanna, F.; Messa, G.; Emanuele, M.; Esposito, A.; Dorofeikova, M.; et al. Pronounced Hyperactivity, Cognitive Dysfunctions, and BDNF Dysregulation in Dopamine Transporter Knock-out Rats. J. Neurosci. 2018, 38, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T.; et al. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 2018, 174, 481–496.e19. [Google Scholar] [CrossRef] [PubMed]
- Patriarchi, T.; Cho, J.R.; Merten, K.; Howe, M.W.; Marley, A.; Xiong, W.H.; Folk, R.W.; Broussard, G.J.; Liang, R.; Jang, M.J.; et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 2018, 360, 6396. [Google Scholar] [CrossRef]
- Morairty, S.R.; Dittrich, L.; Pasumarthi, R.K.; Valladao, D.; Heiss, J.E.; Gerashchenko, D.; Kilduff, T.S. A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc. Natl. Acad. Sci. USA 2013, 110, 20272–20277. [Google Scholar] [CrossRef]
- Dedic, N.; Dworak, H.; Zeni, C.; Rutigliano, G.; Howes, O.D. Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 13185. [Google Scholar] [CrossRef]
- Halff, E.F.; Rutigliano, G.; Garcia-Hidalgo, A.; Howes, O.D. Trace amine-associated receptor 1 (TAAR1) agonism as a new treatment strategy for schizophrenia and related disorders. Trends Neurosci. 2023, 46, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Alnefeesi, Y.; Tamura, J.K.; Lui, L.M.W.; Jawad, M.Y.; Ceban, F.; Ling, S.; Nasri, F.; Rosenblat, J.D.; McIntyre, R.S. Trace amine-associated receptor 1 (TAAR1): Potential application in mood disorders: A systematic review. Neurosci. Biobehav. Rev. 2021, 131, 192–210. [Google Scholar] [CrossRef]
- Dedic, N.; Jones, P.G.; Hopkins, S.C.; Lew, R.; Shao, L.; Campbell, J.E.; Spear, K.L.; Large, T.H.; Campbell, U.C.; Hanania, T.; et al. SEP-363856, a novel psychotropic agent with a unique, non-D2 receptor mechanism of action. J. Pharmacol. Exp. Ther. 2019, 371, 1–14. [Google Scholar] [CrossRef]
- Hopkins, S.C.; Dedic, N.; Koblan, K.S. Effect of TAAR1/5-HT(1A) agonist SEP-363856 on REM sleep in humans. Transl. Psychiatry 2021, 11, 228. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Change Relative to Saline + Vehicle | Change Relative to Saline + RO5263397 | |||||
|---|---|---|---|---|---|---|---|
| D1 Antagonist + Vehicle | D2 Antagonist + Vehicle | D1+D2 Antagonists + Vehicle | Saline + RO5263397 | D1 Antagonist + RO5263397 | D2 Antagonist + RO5263397 | D1+D2 Antagonists + RO5263397 | |
| Wake Time (ZT6-7) | No effect | No effect | No effect | Increased | No effect | Attenuated increase | Reduced increase |
| Wake Time (ZT6-8) | No effect | No effect | No effect | Increased | No effect | Not significant | Reduced increase |
| Cumulative Wake Time (ZT6-11) | Increased | No effect | No effect | Increased | Reduced increase | Reduced increase | Reduced increase |
| Latency to NREM Sleep | No effect | No effect | No effect | Increased | Blocked increase | No effect | No effect |
| NREM Sleep Time (ZT6-7) | No effect | No effect | No effect | Decreased | No effect | Increased | Increased |
| NREM Sleep Time (ZT6-8) | No effect | No effect | No effect | Decreased | No effect | Not significant | Increased |
| Cumulative NREM Time (ZT6-11) | No effect | No effect | No effect | Decreased | Attenuated decrease | Attenuated decrease | Blocked decrease |
| Latency to REM Sleep | No effect | No effect | No effect | Increased | No effect | No effect | No effect |
| REM Sleep Time (ZT6-8) | No effect | No effect | No effect | Decreased | No effect | No effect | No effect |
| Cumulative REM Time (ZT6-11) | No effect | No effect | No effect | Decreased | No effect | No effect | No effect |
| Subcutaneous Body Temperature | Mild ↓: ZT7 | Delayed ↓: ZT10-11 | Prolonged ↓: ZT7-11 | ZT6-7 only | Mild ↓: ZT6-7 | Profound ↓↓: ZT7-9 | Prolonged ↓: ZT7-11 |
| Activity | Reduced at ZT6 only | Reduced throughout | Reduced throughout | Reduced at ZT6 only | Reduced at ZT6 only | Reduced throughout | Reduced throughout |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Heu, J.; Hoener, M.C.; Kilduff, T.S. Wakefulness Induced by TAAR1 Partial Agonism in Mice Is Mediated Through Dopaminergic Neurotransmission. Int. J. Mol. Sci. 2024, 25, 11351. https://doi.org/10.3390/ijms252111351
Park S, Heu J, Hoener MC, Kilduff TS. Wakefulness Induced by TAAR1 Partial Agonism in Mice Is Mediated Through Dopaminergic Neurotransmission. International Journal of Molecular Sciences. 2024; 25(21):11351. https://doi.org/10.3390/ijms252111351
Chicago/Turabian StylePark, Sunmee, Jasmine Heu, Marius C. Hoener, and Thomas S. Kilduff. 2024. "Wakefulness Induced by TAAR1 Partial Agonism in Mice Is Mediated Through Dopaminergic Neurotransmission" International Journal of Molecular Sciences 25, no. 21: 11351. https://doi.org/10.3390/ijms252111351
APA StylePark, S., Heu, J., Hoener, M. C., & Kilduff, T. S. (2024). Wakefulness Induced by TAAR1 Partial Agonism in Mice Is Mediated Through Dopaminergic Neurotransmission. International Journal of Molecular Sciences, 25(21), 11351. https://doi.org/10.3390/ijms252111351