
Differential Proteoglycan Expression in Atherosclerosis Alters Platelet Adhesion and Activation

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
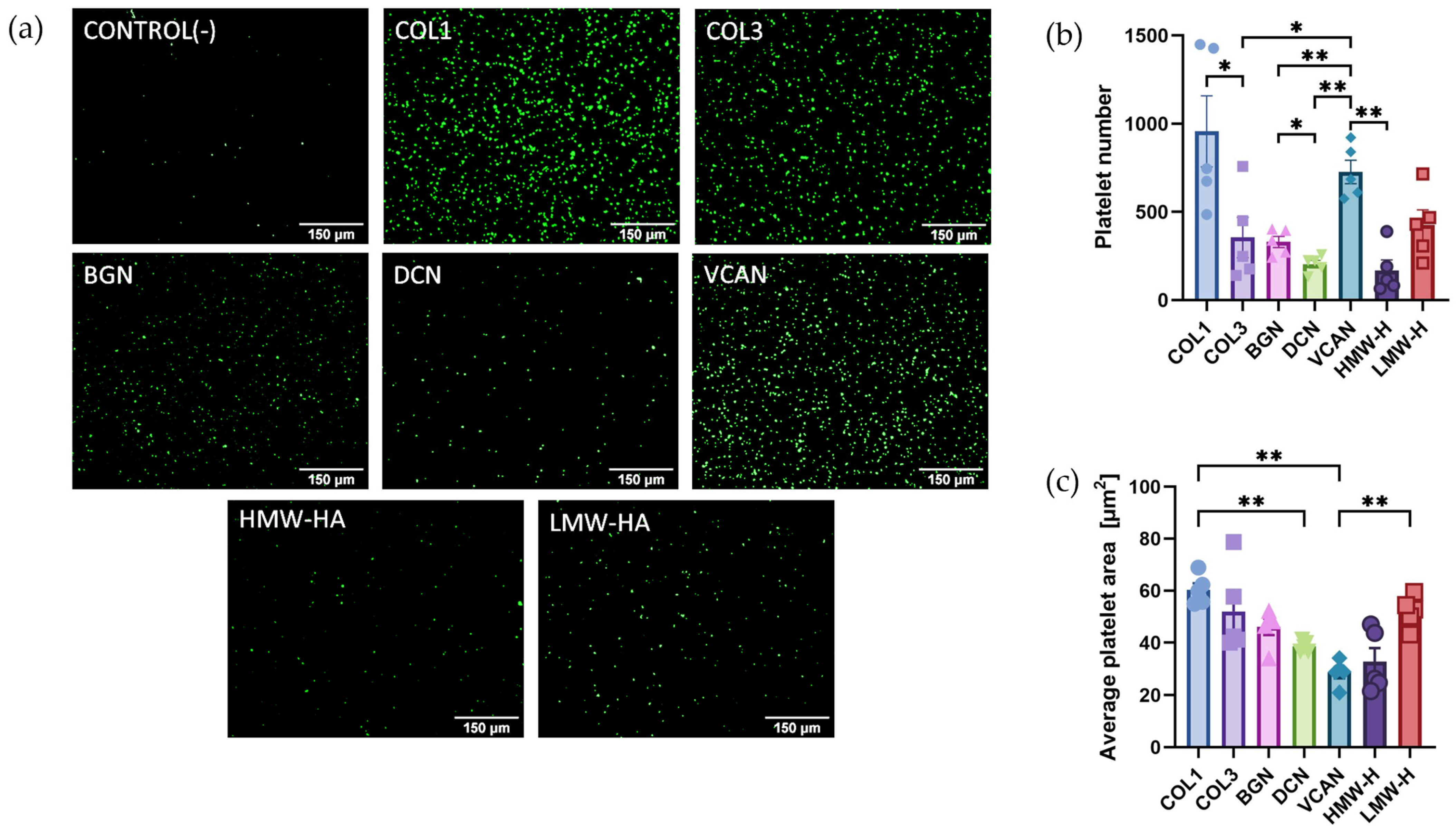
2.1. Recombinant Vascular Proteoglycans Support Platelet Adhesion and Spreading
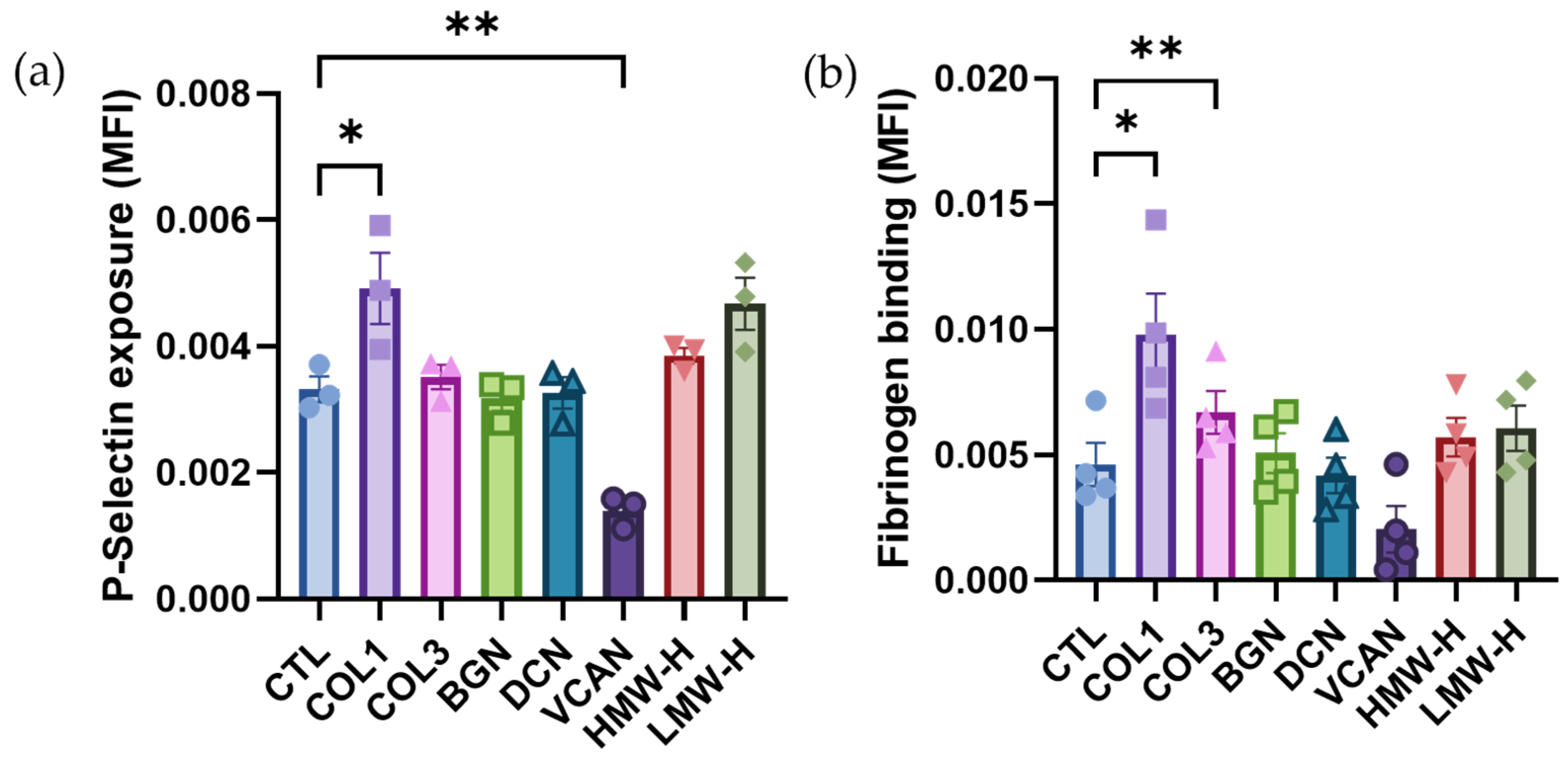
2.2. Vascular Proteoglycans Do Not Stimulate Platelet Activation Responses
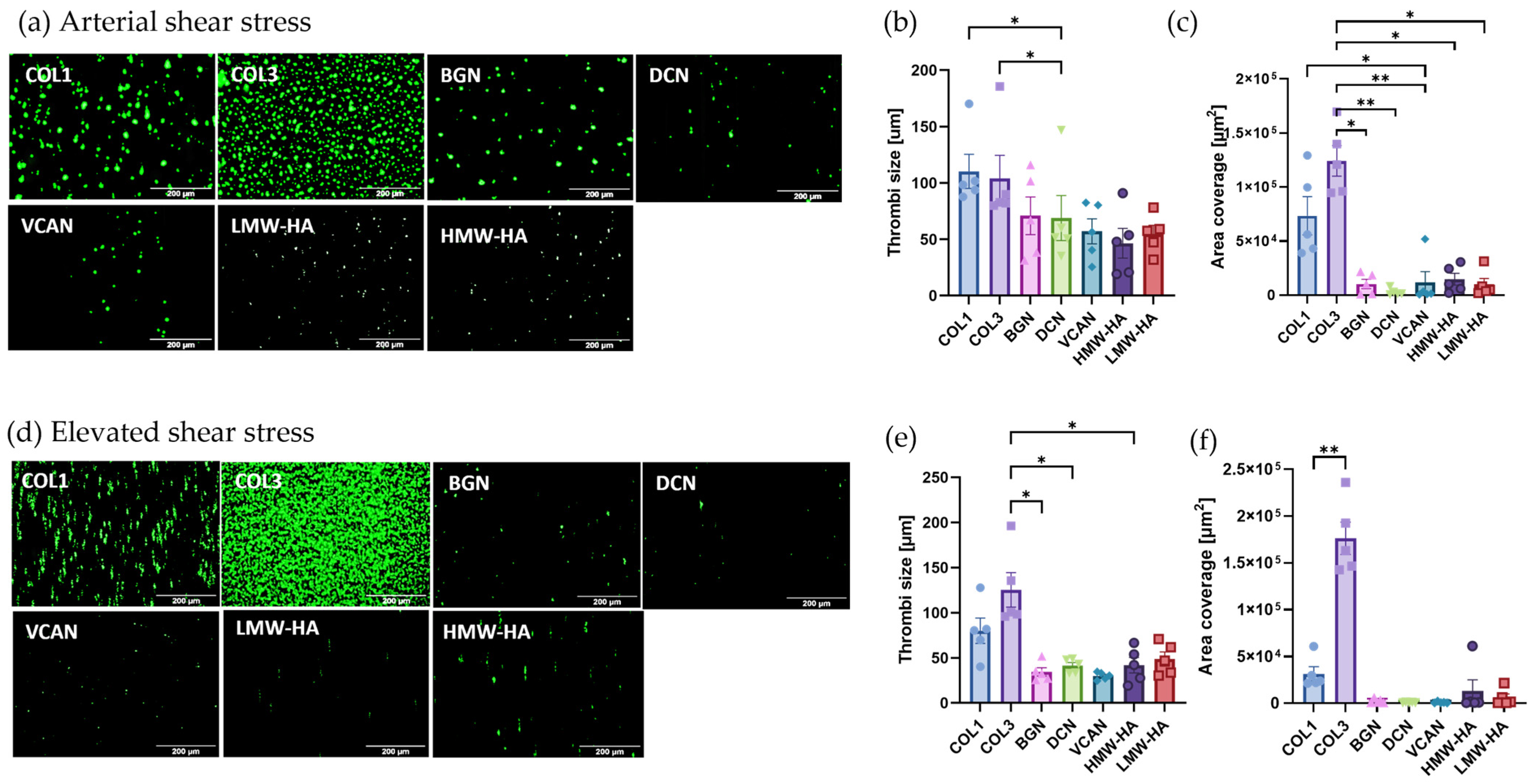
2.3. Vascular Proteoglycans Alone Do Not Support Robust Thrombus Formation under Arterial and Elevated Flow
2.4. Vascular Proteoglycans Regulate Platelet Responses to Collagen
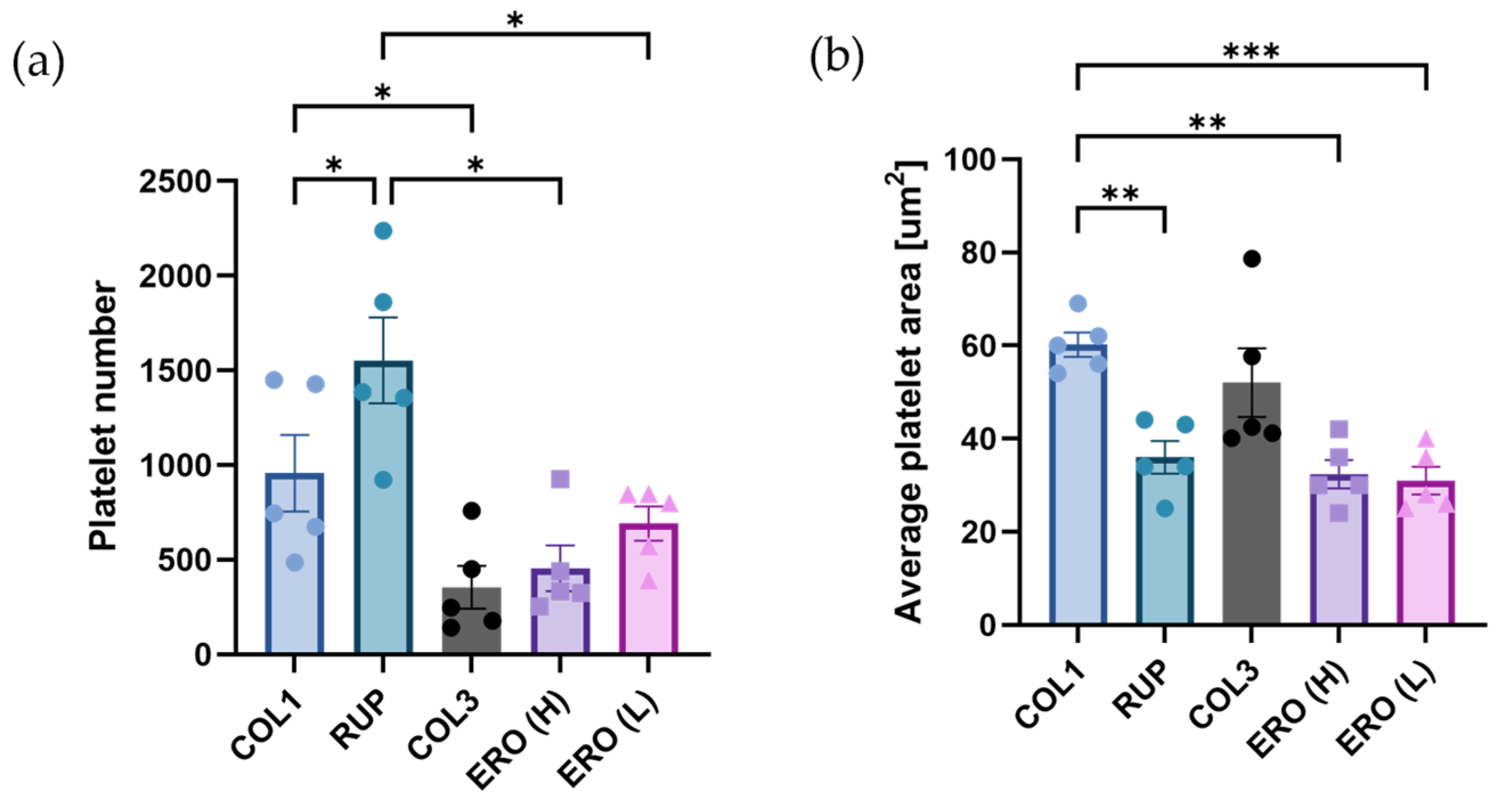
2.5. Composite ‘Rupture’ and ‘Erosion’ Matrices Are Less Thrombogenic than Collagen under Shear
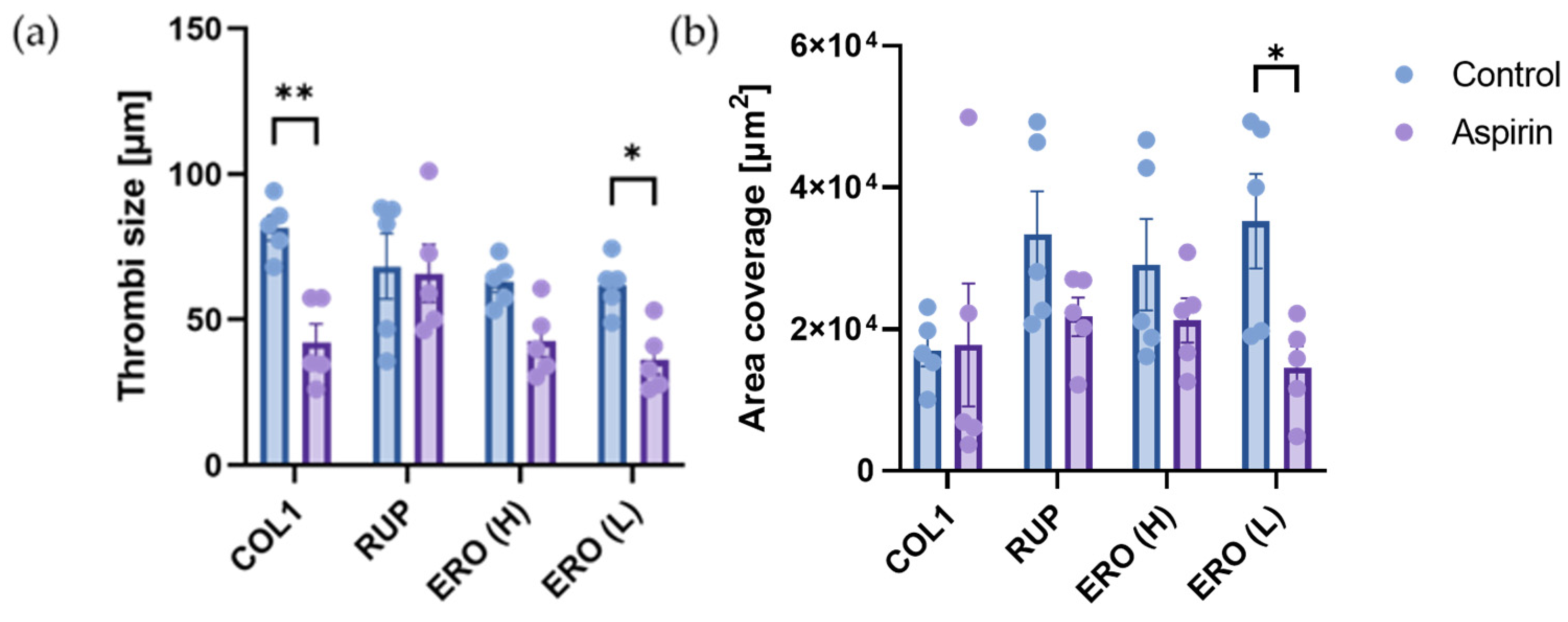
2.6. Aspirin Efficacy Is Altered on Composite ‘Rupture’ and ‘Erosion’ Matrices
2.7. Biglycan and Decorin Exhibit Antithrombotic Effects by Limiting Thrombus Formation on Type I Collagen
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Platelet Preparation
4.3. Platelet Adhesion and Spreading Assays
4.4. Flow Cytometry Analysis of Platelet Activation Markers
4.5. In Vitro Thrombosis Model
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, A.; Unsworth, A.J.; White, S.J.; Jones, S. The Contribution of Vascular Proteoglycans to Atherothrombosis: Clinical Implications. Int. J. Mol. Sci. 2023, 24, 11854. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. The growing complexity of platelet aggregation. Blood 2007, 109, 5087–5095. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Topol, E.J. Scientific and therapeutic advances in antiplatelet therapy. Nat. Rev. Drug Discov. 2003, 2, 15–28. [Google Scholar] [PubMed]
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Wight, T.N.; Virmani, R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: Insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1642–1648. [Google Scholar] [CrossRef]
- Otsuka, F.; Yasuda, S.; Noguchi, T.; Ishibashi-Ueda, H. Pathology of coronary atherosclerosis and thrombosis. Cardiovasc. Diagn. Ther. 2016, 6, 396–408. [Google Scholar] [CrossRef]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef]
- Baaten, C.; Nagy, M.; Bergmeier, W.; Spronk, H.M.H.; van der Meijden, P.E.J. Platelet biology and function: Plaque erosion vs. rupture. Eur. Heart J. 2023, 45, 18–31. [Google Scholar] [CrossRef]
- Wight, T.N. A role for proteoglycans in vascular disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, M.; Petousis, S.; Parthenakis, F. Interaction between platelets and endothelium: From pathophysiology to new therapeutic options. Cardiovasc. Diagn. Ther. 2018, 8, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Burke, A.P.; Wight, T.N.; Virmani, R. The accumulation of specific types of proteoglycans in eroded plaques: A role in coronary thrombosis in the absence of rupture. Curr. Opin. Lipidol. 2004, 15, 575–582. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Newby, A.C.; Johnson, T.W. Endothelial erosion of plaques as a substrate for coronary thrombosis. Thromb. Haemost. 2016, 115, 509–519. [Google Scholar] [PubMed]
- McElroy, M.; Kim, Y.; Niccoli, G.; Vergallo, R.; Langford-Smith, A.; Crea, F.; Gijsen, F.; Johnson, T.; Keshmiri, A.; White, S.J. Identification of the haemodynamic environment permissive for plaque erosion. Sci. Rep. 2021, 11, 7253. [Google Scholar] [CrossRef]
- Mazzucato, M.; Cozzi, M.R.; Pradella, P.; Perissinotto, D.; Malmstrom, A.; Morgelin, M.; Spessotto, P.; Colombatti, A.; De Marco, L.; Perris, R. Vascular PG-M/versican variants promote platelet adhesion at low shear rates and cooperate with collagens to induce aggregation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- de Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef] [PubMed]
- Koshiishi, I.; Shizari, M.; Underhill, C.B. CD44 can mediate the adhesion of platelets to hyaluronan. Blood 1994, 84, 390–396. [Google Scholar] [CrossRef]
- Svensson Holm, A.C.; Bengtsson, T.; Grenegard, M.; Lindstrom, E.G. Hyaluronic acid influence on platelet-induced airway smooth muscle cell proliferation. Exp. Cell Res. 2012, 318, 632–640. [Google Scholar] [CrossRef]
- Wu, Y.J.; La Pierre, D.P.; Wu, J.; Yee, A.J.; Yang, B.B. The interaction of versican with its binding partners. Cell Res. 2005, 15, 483–494. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef]
- Savage, B.; Ginsberg, M.H.; Ruggeri, Z.M. Influence of fibrillar collagen structure on the mechanisms of platelet thrombus formation under flow. Blood 1999, 94, 2704–2715. [Google Scholar] [CrossRef]
- Machha, V.R.; Tischer, A.; Moon-Tasson, L.; Auton, M. The Von Willebrand Factor A1-Collagen III Interaction Is Independent of Conformation and Type 2 Von Willebrand Disease Phenotype. J. Mol. Biol. 2017, 429, 32–47. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Fahed, A.C.; Jang, I.K. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 2021, 18, 724–734. [Google Scholar] [CrossRef]
- Jia, H.; Abtahian, F.; Aguirre, A.D.; Lee, S.; Chia, S.; Lowe, H.; Kato, K.; Yonetsu, T.; Vergallo, R.; Hu, S.; et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J. Am. Coll. Cardiol. 2013, 62, 1748–1758. [Google Scholar] [CrossRef]
- Paderi, J.E.; Stuart, K.; Sturek, M.; Park, K.; Panitch, A. The inhibition of platelet adhesion and activation on collagen during balloon angioplasty by collagen-binding peptidoglycans. Biomaterials 2011, 32, 2516–2523. [Google Scholar] [CrossRef]
- Drysdale, A.; Zaabalawi, A.; Jones, S. Modelling arterial thrombus formation in vitro. Curr. Opin. Hematol. 2023, 31, 16–23. [Google Scholar] [CrossRef]
- Jones, S.; Tucker, K.L.; Sage, T.; Kaiser, W.J.; Barrett, N.E.; Lowry, P.J.; Zimmer, A.; Hunt, S.P.; Emerson, M.; Gibbins, J.M. Peripheral tachykinins and the neurokinin receptor NK1 are required for platelet thrombus formation. Blood 2008, 111, 605–612. [Google Scholar] [CrossRef]
- Tucker, K.L.; Sage, T.; Stevens, J.M.; Jordan, P.A.; Jones, S.; Barrett, N.E.; St-Arnaud, R.; Frampton, J.; Dedhar, S.; Gibbins, J.M. A dual role for integrin-linked kinase in platelets: Regulating integrin function and alpha-granule secretion. Blood 2008, 112, 4523–4531. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Bye, A.P.; Sage, T.; Gaspar, R.S.; Eaton, N.; Drew, C.; Stainer, A.; Kriek, N.; Volberding, P.J.; Hutchinson, J.L.; et al. Antiplatelet properties of Pim kinase inhibition are mediated through disruption of thromboxane A2 receptor signaling. Haematologica 2021, 106, 1968–1978. [Google Scholar] [CrossRef]
- Kriek, N.; Nock, S.H.; Sage, T.; Khalifa, B.; Bye, A.P.; Mitchell, J.L.; Thomson, S.; McLaughlin, M.G.; Jones, S.; Gibbins, J.M.; et al. Cucurbitacins Elicit Anti-Platelet Activity via Perturbation of the Cytoskeleton and Integrin Function. Thromb. Haemost. 2022, 122, 1115–1129. [Google Scholar] [CrossRef]
- Sokolowska, M.; Chen, L.Y.; Eberlein, M.; Martinez-Anton, A.; Liu, Y.; Alsaaty, S.; Qi, H.Y.; Logun, C.; Horton, M.; Shelhamer, J.H. Low molecular weight hyaluronan activates cytosolic phospholipase A2alpha and eicosanoid production in monocytes and macrophages. J. Biol. Chem. 2014, 289, 4470–4488. [Google Scholar] [CrossRef]
- Antithrombotic Trialists, C.; Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; et al. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009, 373, 1849–1860. [Google Scholar]
- Cosemans, J.M.; Kuijpers, M.J.; Lecut, C.; Loubele, S.T.; Heeneman, S.; Jandrot-Perrus, M.; Heemskerk, J.W. Contribution of platelet glycoprotein VI to the thrombogenic effect of collagens in fibrous atherosclerotic lesions. Atherosclerosis 2005, 181, 19–27. [Google Scholar] [CrossRef]
- Nieswandt, B.; Pleines, I.; Bender, M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 92–104. [Google Scholar] [CrossRef]
- Martins Lima, A.; Martins Cavaco, A.C.; Fraga-Silva, R.A.; Eble, J.A.; Stergiopulos, N. From Patients to Platelets and Back Again: Pharmacological Approaches to Glycoprotein VI, a Thrilling Antithrombotic Target with Minor Bleeding Risks. Thromb. Haemost. 2019, 119, 1720–1739. [Google Scholar] [CrossRef]
- Mayer, K.; Hein-Rothweiler, R.; Schupke, S.; Janisch, M.; Bernlochner, I.; Ndrepepa, G.; Sibbing, D.; Gori, T.; Borst, O.; Holdenrieder, S.; et al. Efficacy and Safety of Revacept, a Novel Lesion-Directed Competitive Antagonist to Platelet Glycoprotein VI, in Patients Undergoing Elective Percutaneous Coronary Intervention for Stable Ischemic Heart Disease: The Randomized, Double-blind, Placebo-Controlled ISAR-PLASTER Phase 2 Trial. JAMA Cardiol. 2021, 6, 753–761. [Google Scholar]
- Billiald, P.; Slater, A.; Welin, M.; Clark, J.C.; Loyau, S.; Pugniere, M.; Jiacomini, I.G.; Rose, N.; Lebozec, K.; Toledano, E.; et al. Targeting platelet GPVI with glenzocimab: A novel mechanism for inhibition. Blood Adv. 2023, 7, 1258–1268. [Google Scholar] [CrossRef]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef]
- Schonherr, E.; Witsch-Prehm, P.; Harrach, B.; Robenek, H.; Rauterberg, J.; Kresse, H. Interaction of biglycan with type I collagen. J. Biol. Chem. 1995, 270, 2776–2783. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, E.; Hausser, H.; Beavan, L.; Kresse, H. Decorin-type I collagen interaction. Presence of separate core protein-binding domains. J. Biol. Chem. 1995, 270, 8877–8883. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Paderi, J.E.; Sturek, M.; Panitch, A. Decorin mimic inhibits vascular smooth muscle proliferation and migration. PLoS ONE 2013, 8, e82456. [Google Scholar] [CrossRef]
- Grandoch, M.; Kohlmorgen, C.; Melchior-Becker, A.; Feldmann, K.; Homann, S.; Muller, J.; Kiene, L.S.; Zeng-Brouwers, J.; Schmitz, F.; Nagy, N.; et al. Loss of Biglycan Enhances Thrombin Generation in Apolipoprotein E-Deficient Mice: Implications for Inflammation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e41–e50. [Google Scholar] [CrossRef] [PubMed]
- Provenzale, I.; De Simone, I.; Gibbins, J.M.; Heemskerk, J.W.M.; van der Meijden, P.E.J.; Jones, C.I. Regulation of Glycoprotein VI-Dependent Platelet Activation and Thrombus Formation by Heparan Sulfate Proteoglycan Perlecan. Int. J. Mol. Sci. 2023, 24, 13352. [Google Scholar] [CrossRef]
- Walimbe, T.; Panitch, A. Proteoglycans in Biomedicine: Resurgence of an Underexploited Class of ECM Molecules. Front. Pharmacol. 2019, 10, 1661. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drysdale, A.; Blanco-Lopez, M.; White, S.J.; Unsworth, A.J.; Jones, S. Differential Proteoglycan Expression in Atherosclerosis Alters Platelet Adhesion and Activation. Int. J. Mol. Sci. 2024, 25, 950. https://doi.org/10.3390/ijms25020950
Drysdale A, Blanco-Lopez M, White SJ, Unsworth AJ, Jones S. Differential Proteoglycan Expression in Atherosclerosis Alters Platelet Adhesion and Activation. International Journal of Molecular Sciences. 2024; 25(2):950. https://doi.org/10.3390/ijms25020950
Chicago/Turabian StyleDrysdale, Amelia, Maria Blanco-Lopez, Stephen J. White, Amanda J. Unsworth, and Sarah Jones. 2024. "Differential Proteoglycan Expression in Atherosclerosis Alters Platelet Adhesion and Activation" International Journal of Molecular Sciences 25, no. 2: 950. https://doi.org/10.3390/ijms25020950
APA StyleDrysdale, A., Blanco-Lopez, M., White, S. J., Unsworth, A. J., & Jones, S. (2024). Differential Proteoglycan Expression in Atherosclerosis Alters Platelet Adhesion and Activation. International Journal of Molecular Sciences, 25(2), 950. https://doi.org/10.3390/ijms25020950