
Establishment and Validation of Novel Prognostic Subtypes in Hepatocellular Carcinoma Based on Bile Acid Metabolism Gene Signatures Using Bulk and Single-Cell RNA-Seq Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
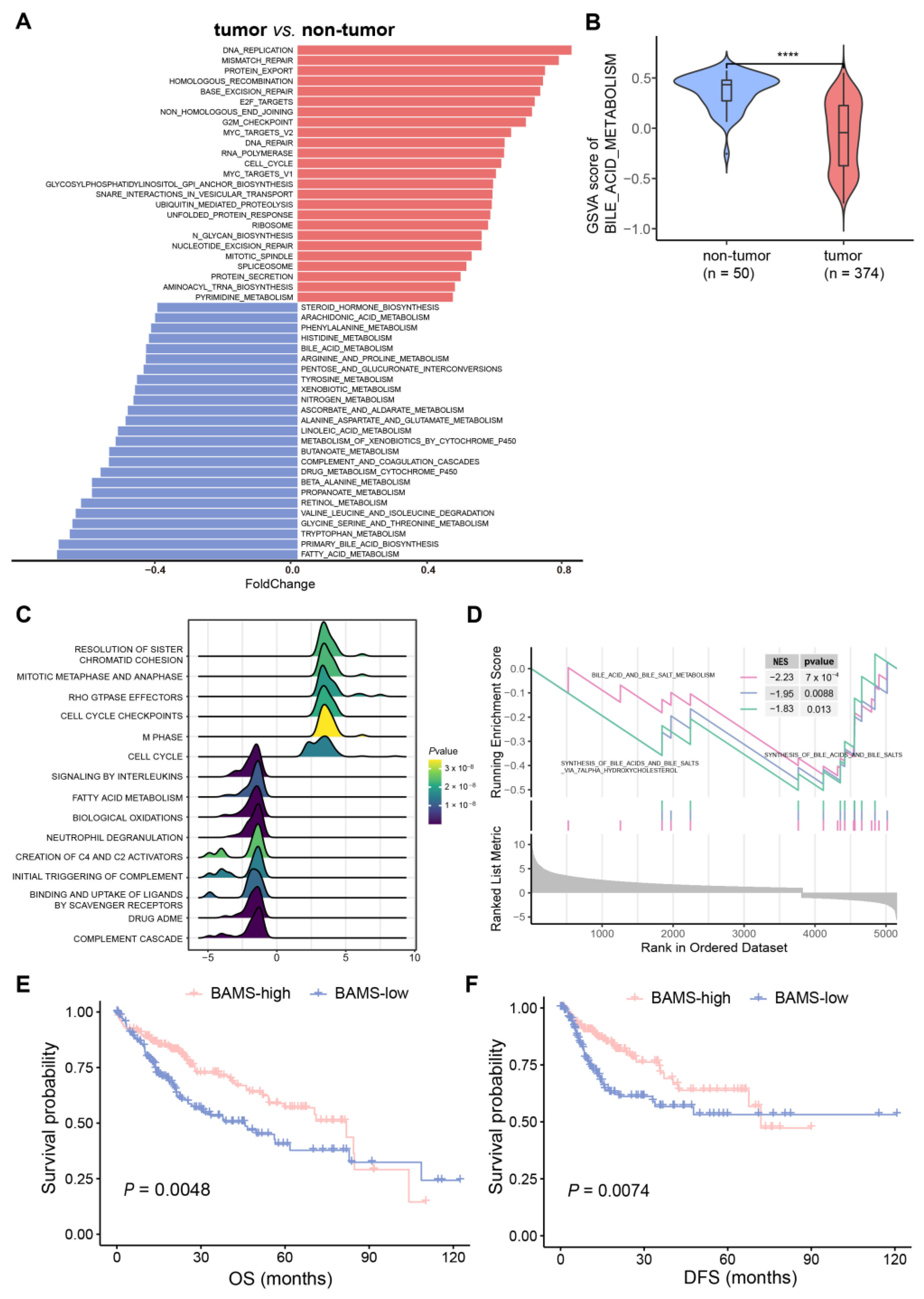
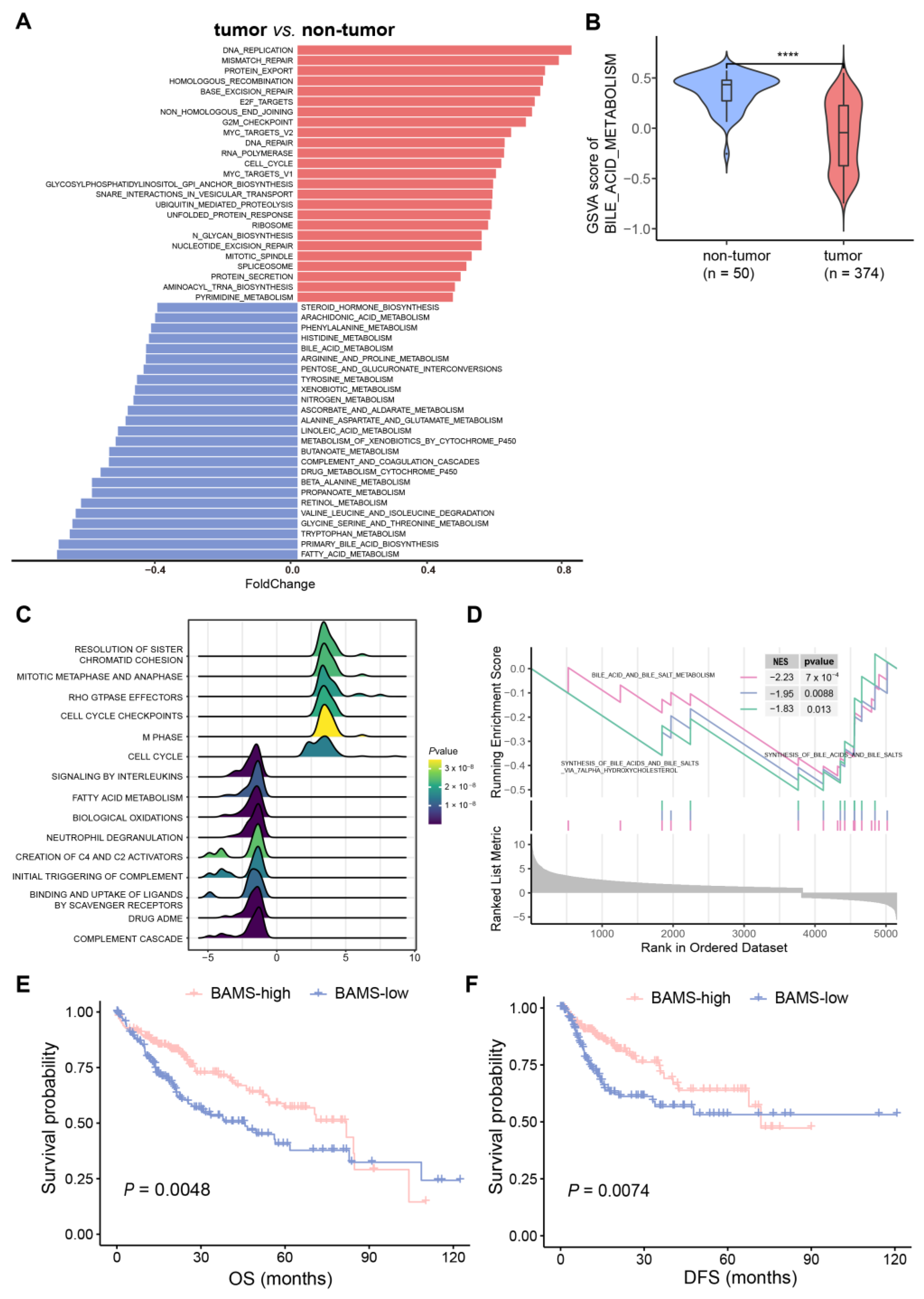
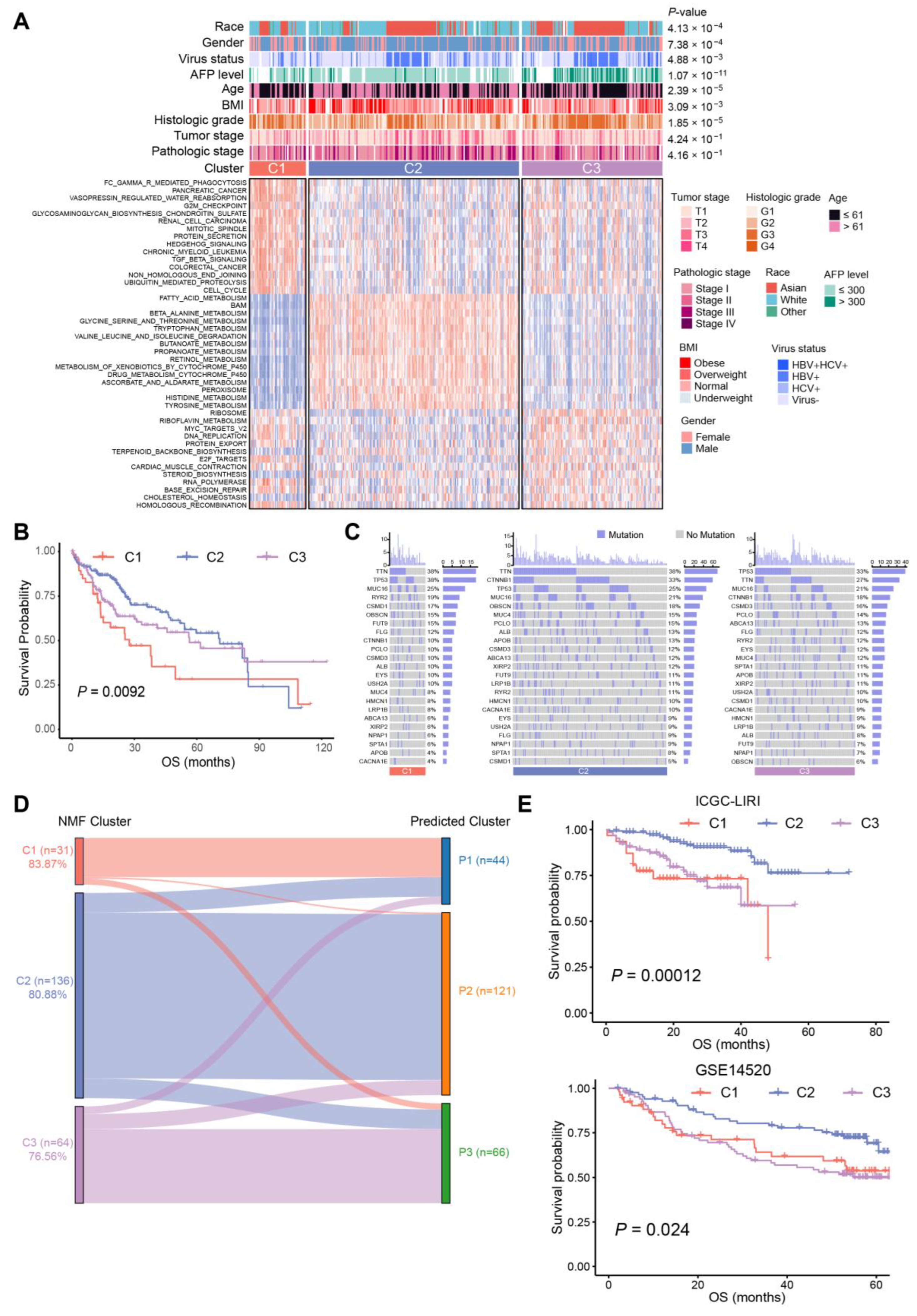
2.1. Altered BA Metabolism Associated with Cancer Biological Pathways, Clinical Prognosis, and Advanced Progression
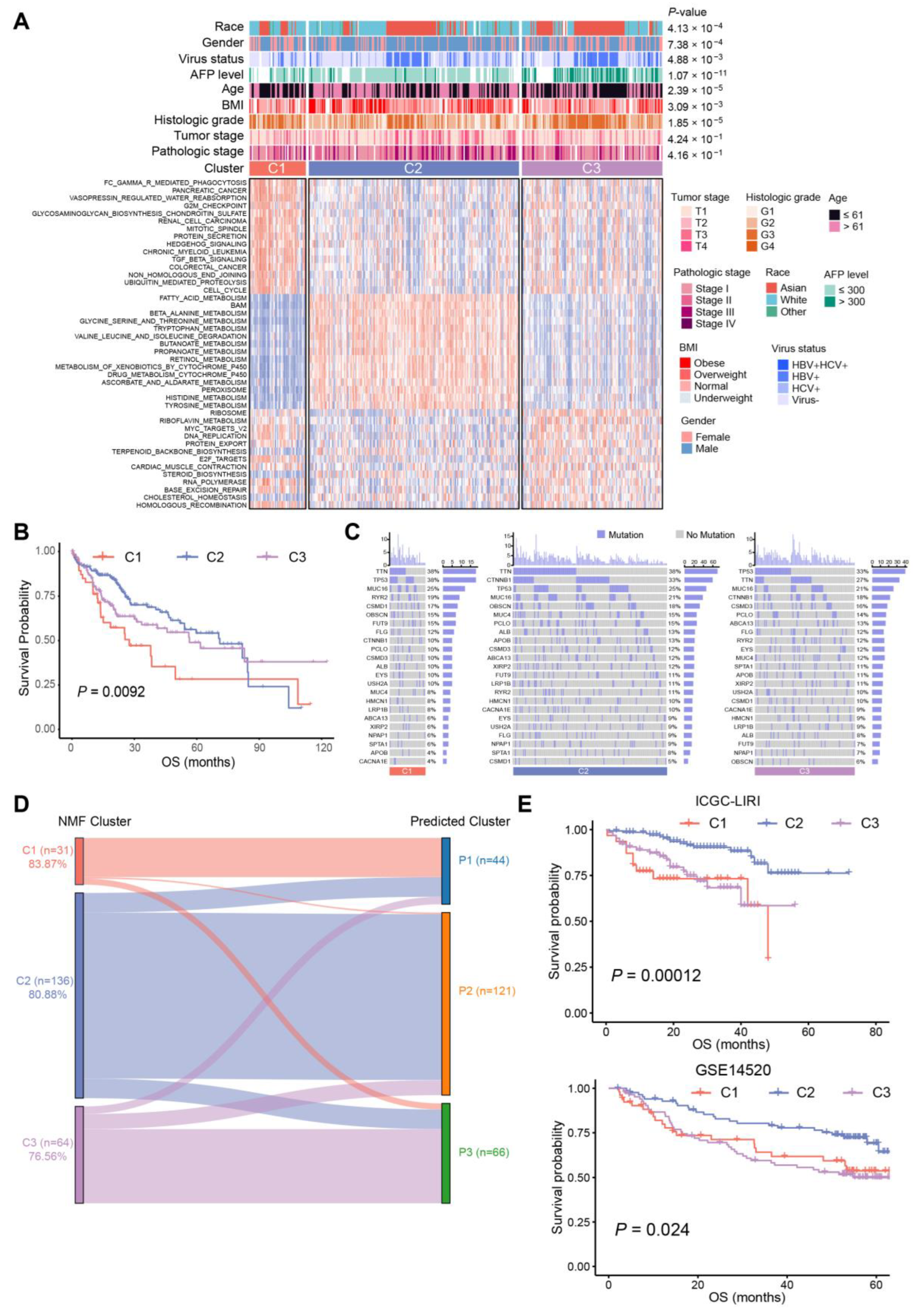
2.2. Three Distinct Subtypes in HCC Identified Based on BA Metabolism-Related Genes Using NMF Methods
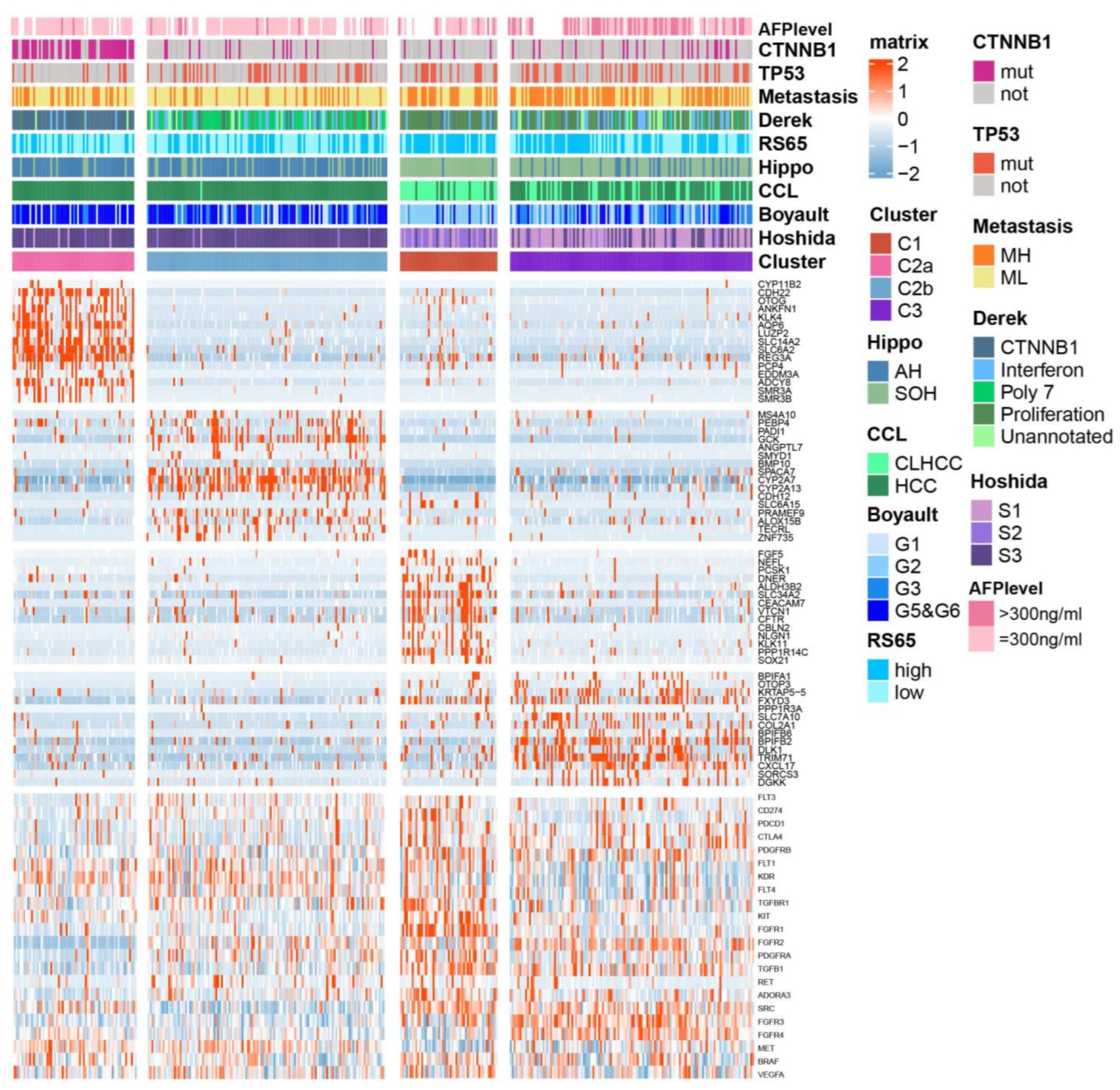
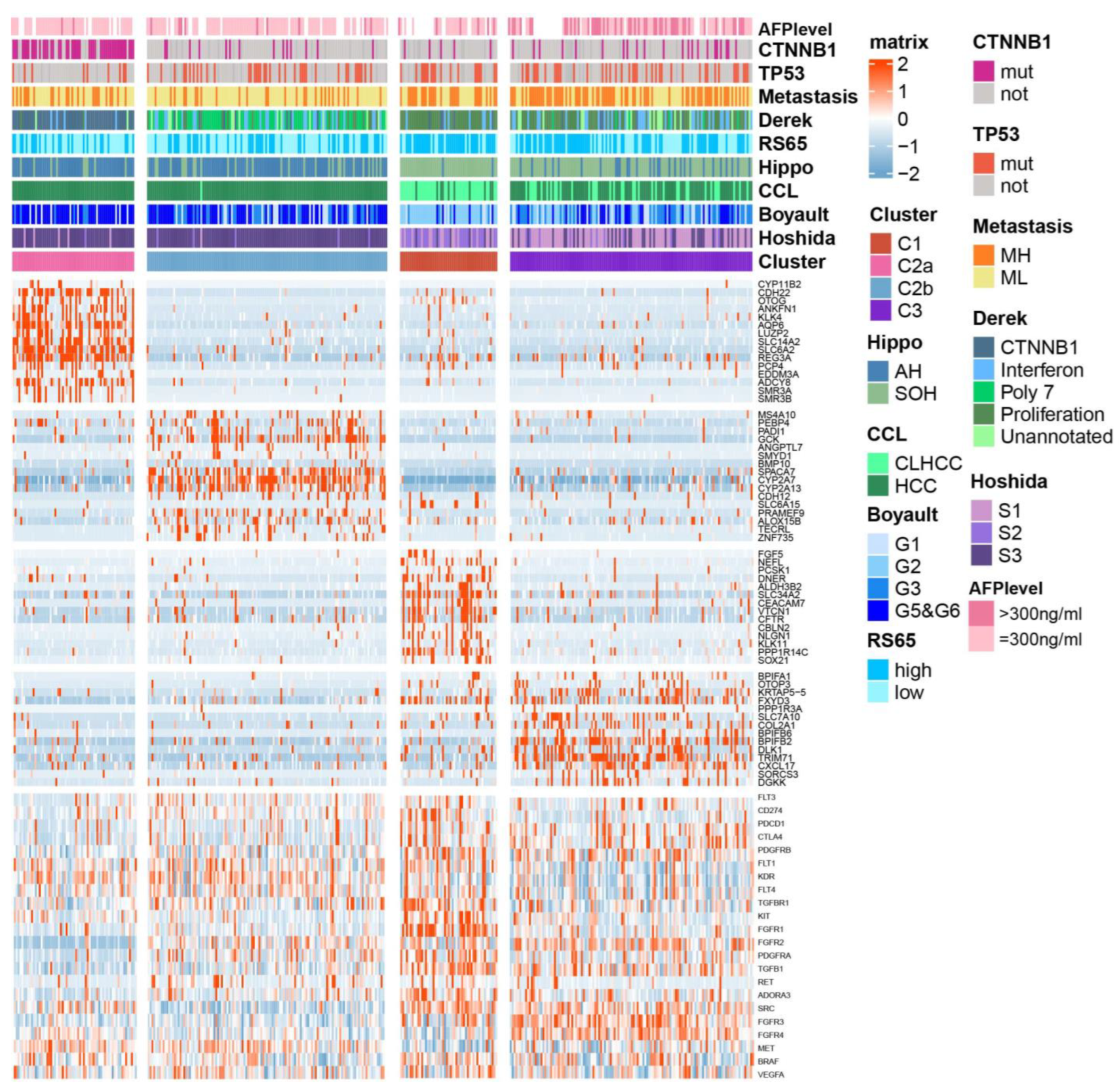
2.3. Association between BA Metabolism-Related Subtypes and Previous Subtypes in HCC
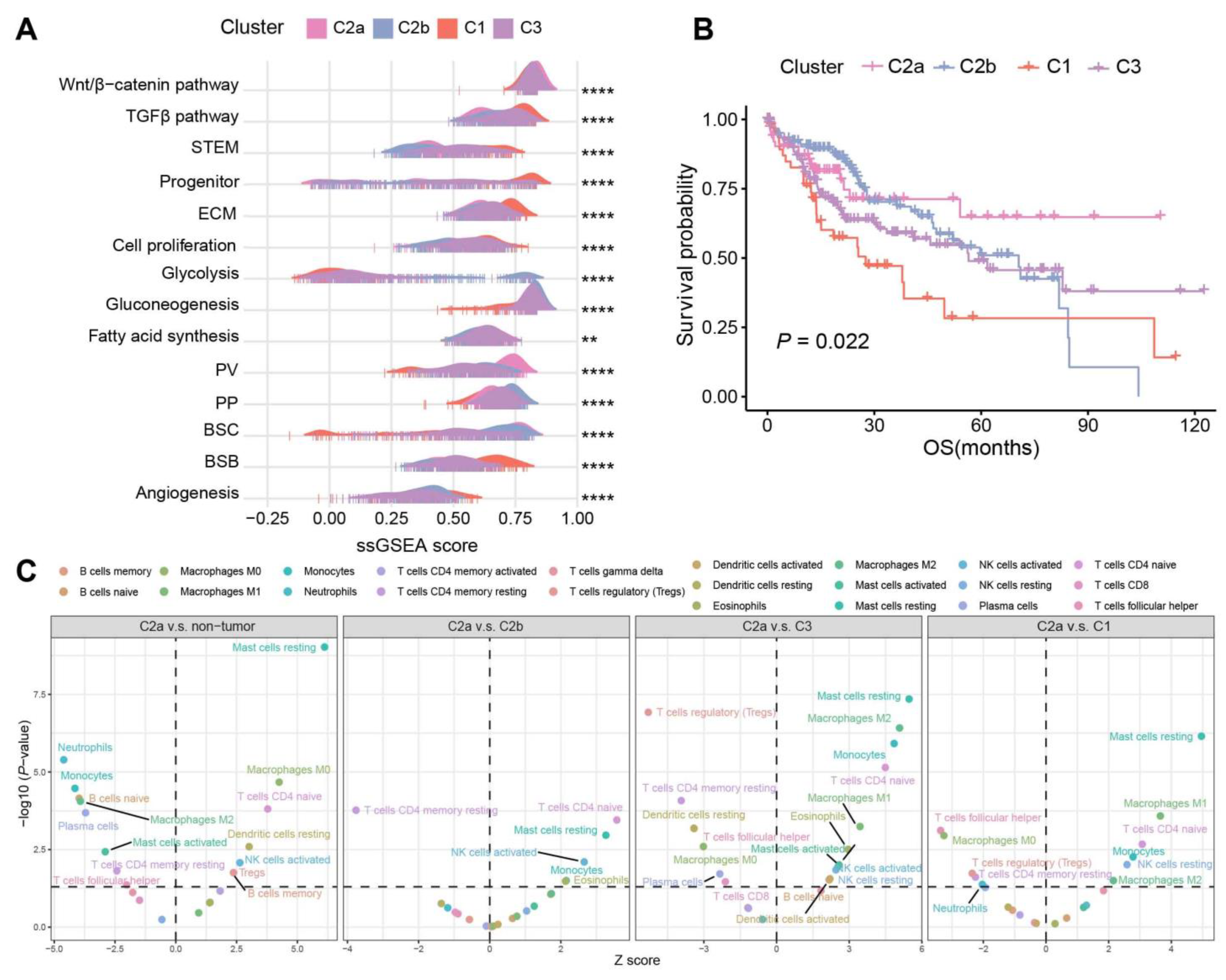
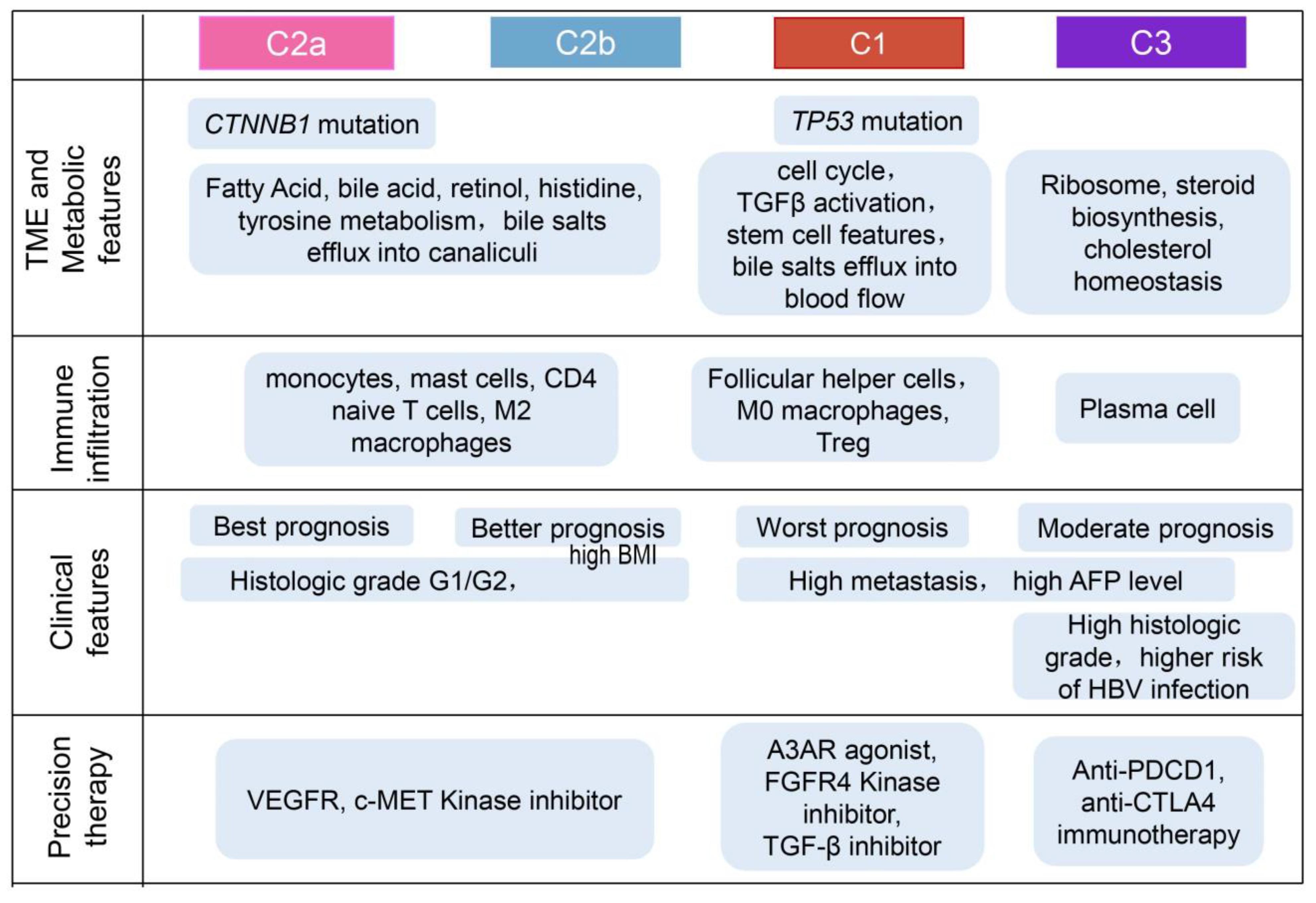
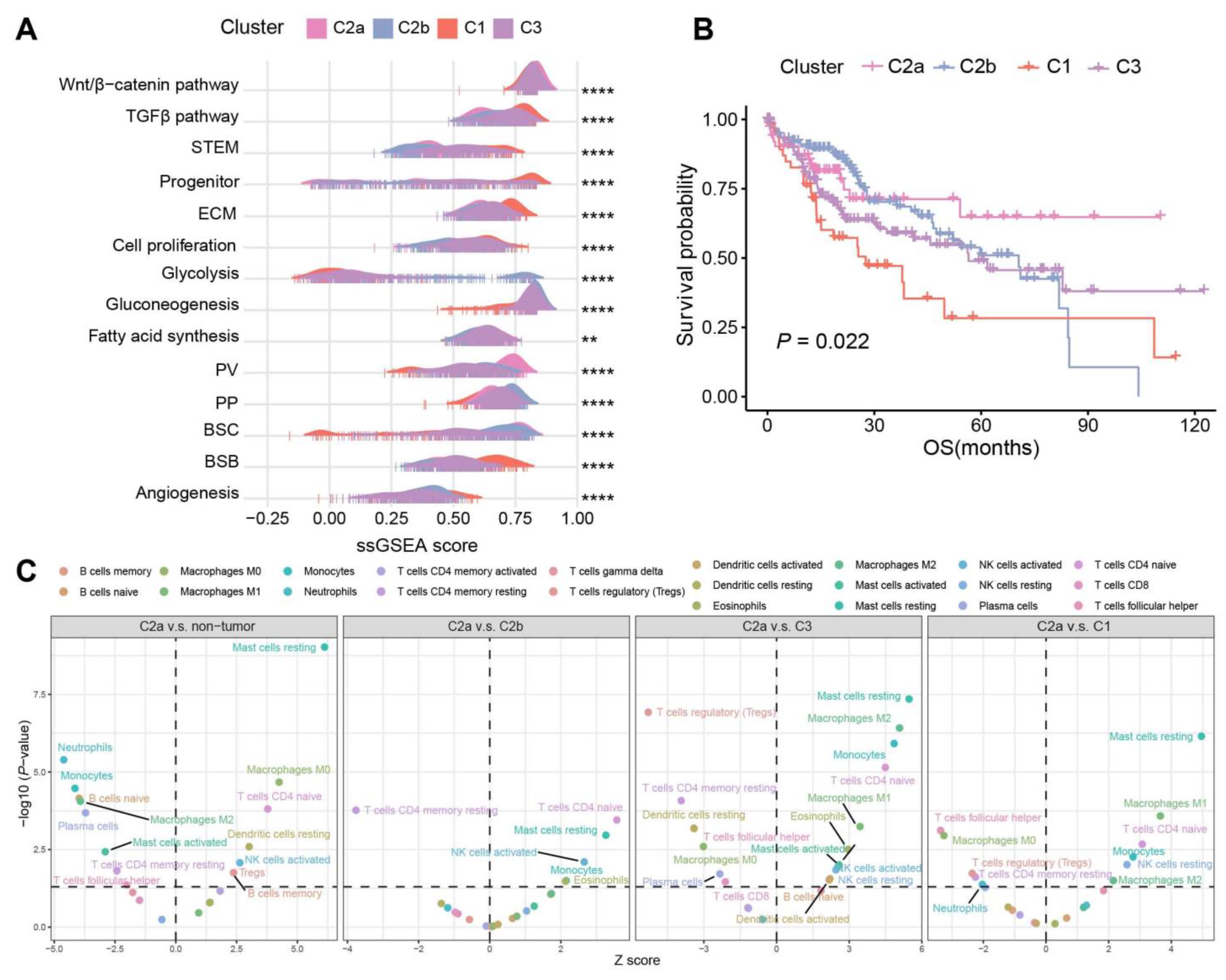
2.4. Biological Characteristics and Immune Infiltration Level between Different Subtypes
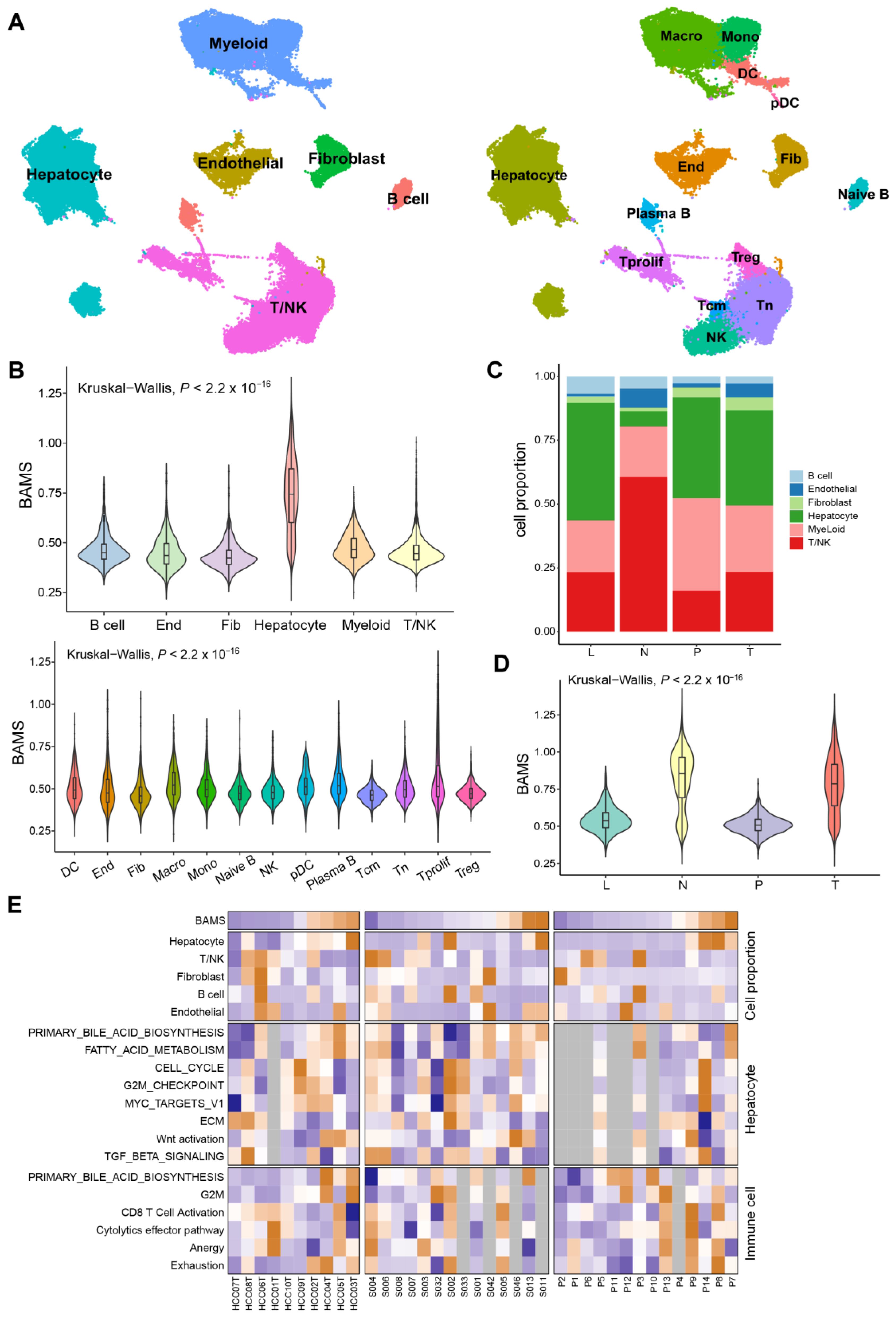
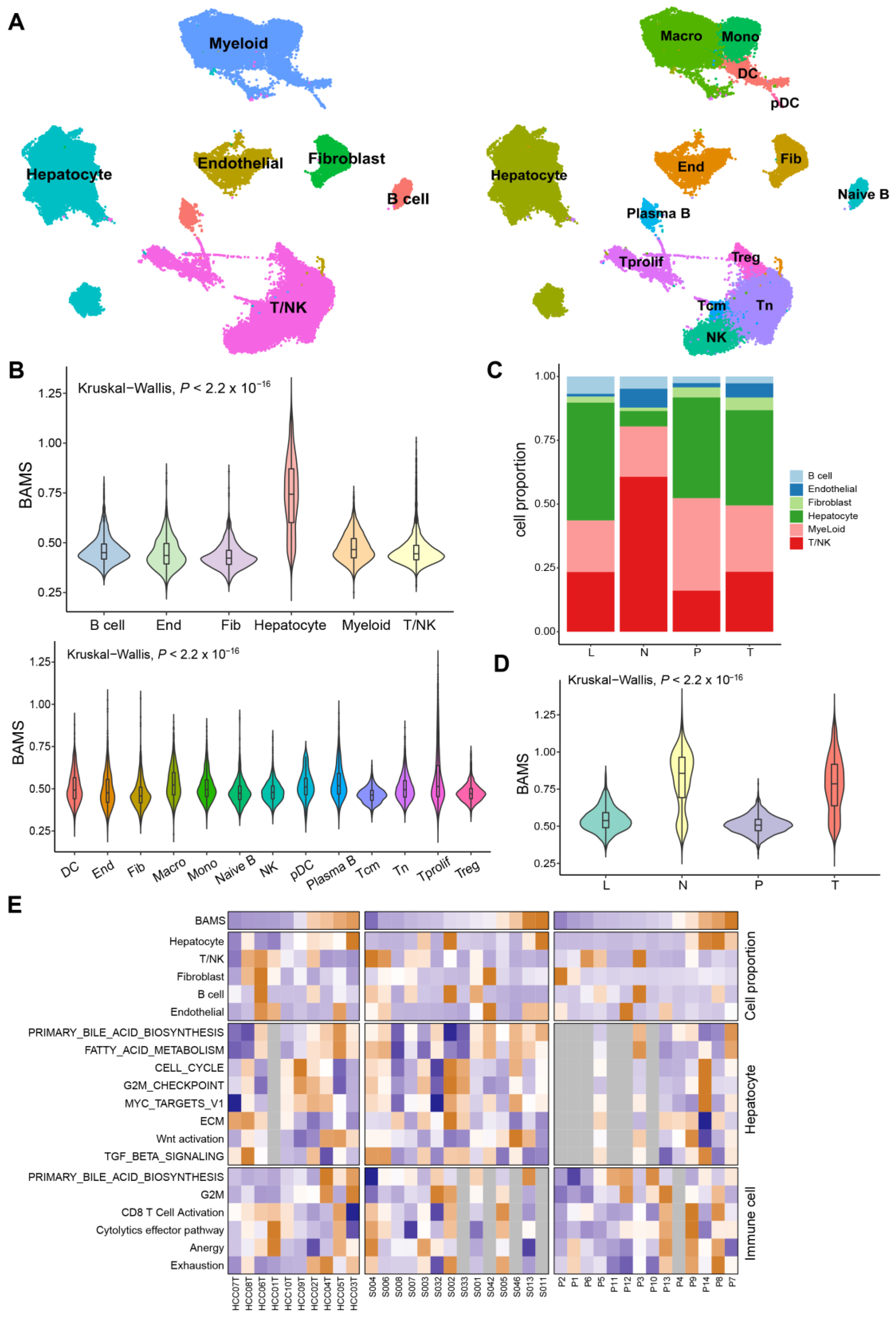
2.5. BA Metabolism in the Level of Single-Cell Transcriptome Data
2.6. Association between BA Metabolism-Related Genes and Cell Cycle-Related Genes
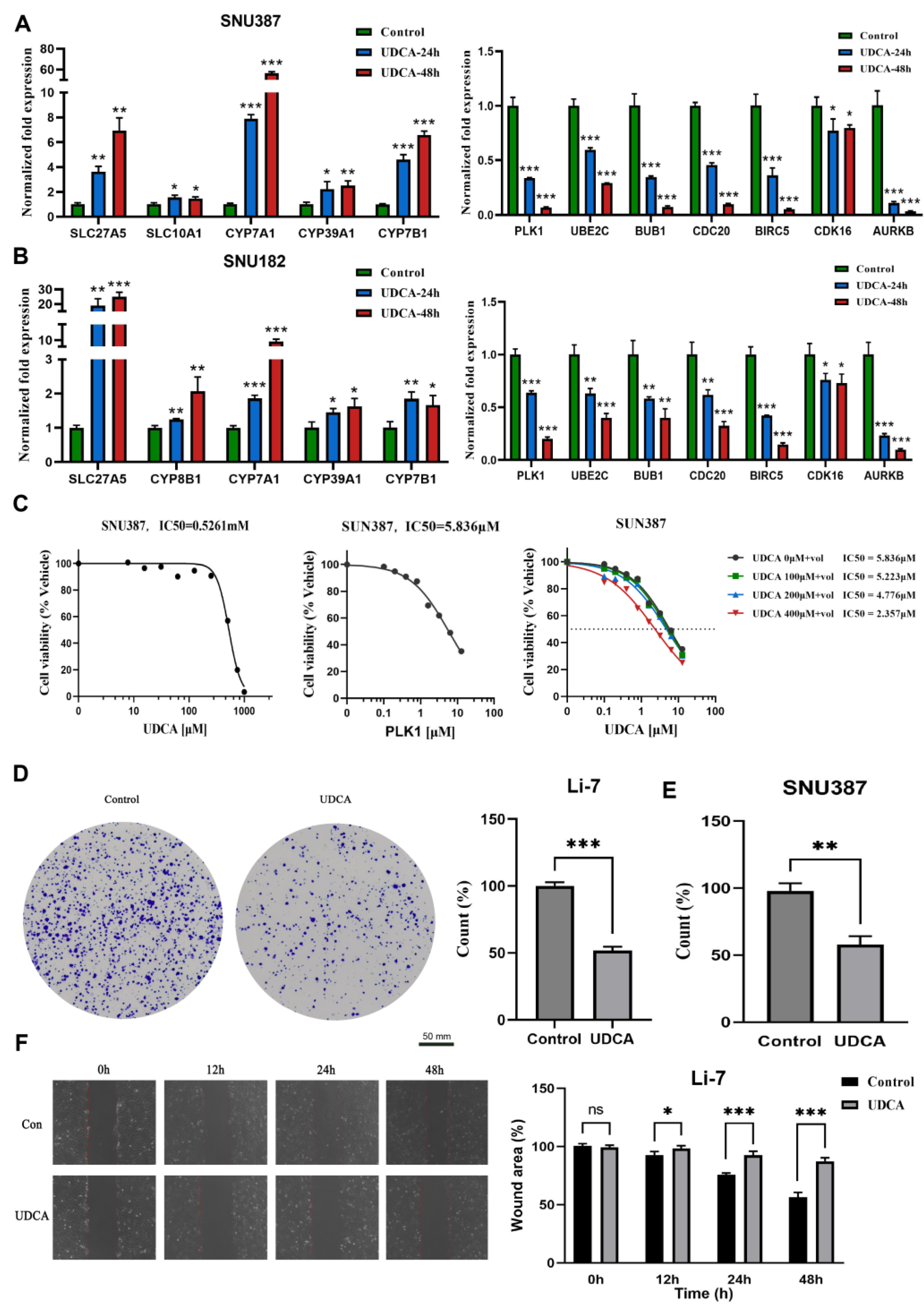
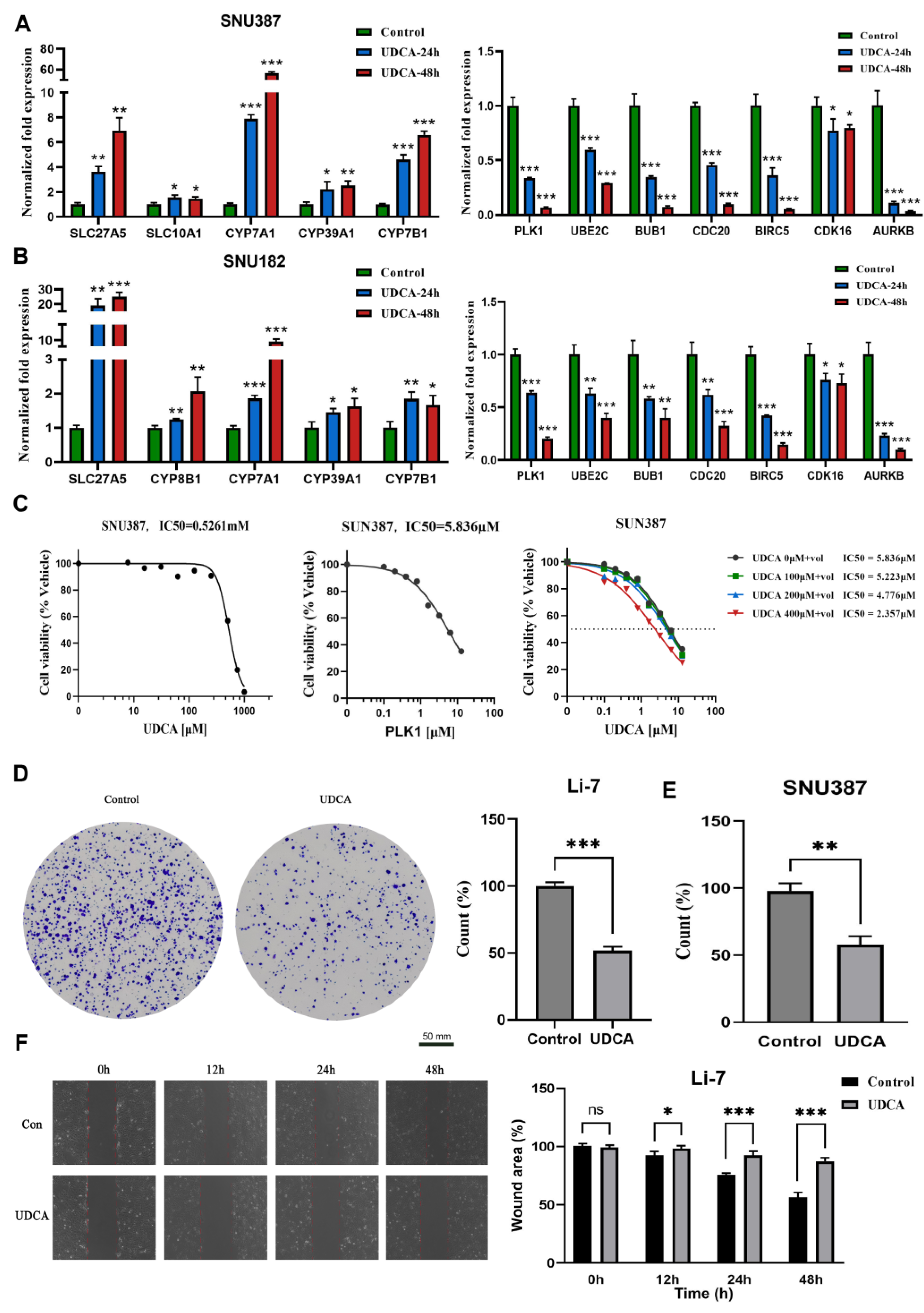
2.7. UDCA Inhibits Cell Cycle-Related Genes and Markedly Affects Cancer Cell Phenotype
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Data Processing
4.2. Gene Set Variation Analysis
4.3. Identification of HCC Subtypes
4.4. Characterization of HCC Subtypes by Previous Molecular Signatures
4.5. Single-Cell RNA Sequencing (scRNA-Seq) Analysis
4.6. Reagents
4.7. Cell Culture
4.8. Cell Counting Kit-8 (CCK-8) Assay
4.9. Migration Assays
4.10. Colony Formation Assay
4.11. qRT-PCR Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, L.; Wang, X.; Tang, Y.; Huang, S.; Hu, C.A.; Teng, Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J. Exp. Clin. Cancer Res. 2017, 36, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.; Chan, K.E.; Wong, Z.Y.; Tan, C.; Tan, B.; Lim, W.H.; Tan, D.; Tang, A.; Tay, P.; Xiao, J.; et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Åberg, F.; Byrne, C.D.; Pirola, C.J.; Männistö, V.; Sookoian, S. Alcohol consumption and metabolic syndrome: Clinical and epidemiological impact on liver disease. J. Hepatol. 2023, 78, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.A.; Patil, R.; Harrison, S.A. NAFLD-related hepatocellular carcinoma: The growing challenge. Hepatology 2023, 77, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Chiang, J.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Tonin, F.; Arends, I. Latest development in the synthesis of ursodeoxycholic acid (UDCA): A critical review. Beilstein J. Org. Chem. 2018, 14, 470–483. [Google Scholar] [CrossRef]
- Satriano, L.; Lewinska, M.; Rodrigues, P.M.; Banales, J.M.; Andersen, J.B. Metabolic rearrangements in primary liver cancers: Cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Iñarrairaegui, M.; Arbelaiz, A.; Milkiewicz, P.; Muntané, J.; Muñoz-Bellvis, L.; La Casta, A.; Gonzalez, L.M.; Arretxe, E.; Alonso, C.; et al. Serum Metabolites as Diagnostic Biomarkers for Cholangiocarcinoma, Hepatocellular Carcinoma, and Primary Sclerosing Cholangitis. Hepatology 2019, 70, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, L.; Yin, P.; Wang, J.; Lu, X.; Wang, X.; Chen, J.; Lin, X.; Xu, G. A weighted relative difference accumulation algorithm for dynamic metabolomics data: Long-term elevated bile acids are risk factors for hepatocellular carcinoma. Sci. Rep. 2015, 5, 8984. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Yin, P.; Hua, R.; Tan, Y.; Li, Z.; Qiu, G.; Yin, Z.; Xie, X.; Wang, X.; Chen, W.; et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 2018, 67, 662–675. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Sohn, B.H.; Shim, J.J.; Kim, S.B.; Jang, K.Y.; Kim, S.M.; Kim, J.H.; Hwang, J.E.; Jang, H.J.; Lee, H.S.; Kim, S.C.; et al. Inactivation of Hippo Pathway Is Significantly Associated with Poor Prognosis in Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 1256–1264. [Google Scholar] [CrossRef]
- Woo, H.G.; Lee, J.H.; Yoon, J.H.; Kim, C.Y.; Lee, H.S.; Jang, J.J.; Yi, N.J.; Suh, K.S.; Lee, K.U.; Park, E.S.; et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010, 70, 3034–3041. [Google Scholar] [CrossRef]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Roessler, S.; Jia, H.L.; Budhu, A.; Forgues, M.; Ye, Q.H.; Lee, J.S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.Y.; Qin, L.X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Leem, S.H.; Chu, I.S.; Park, Y.Y.; Kim, S.C.; Kim, S.B.; Park, E.S.; Lim, J.Y.; Heo, J.; Kim, Y.J.; et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology 2012, 55, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Désert, R.; Rohart, F.; Canal, F.; Sicard, M.; Desille, M.; Renaud, S.; Turlin, B.; Bellaud, P.; Perret, C.; Clément, B.; et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017, 66, 1502–1518. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Marschall, H.U.; Wagner, M.; Zollner, G.; Fickert, P.; Silbert, D.; Gustafsson, U.; Sahlin, S.; Trauner, M. Combined rifampicin and ursodeoxycholic acid treatment does not amplify rifampicin effects on hepatic detoxification and transport systems in humans. Digestion 2012, 86, 244–249. [Google Scholar] [CrossRef]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef]
- Režen, T.; Rozman, D.; Kovács, T.; Kovács, P.; Sipos, A.; Bai, P.; Mikó, E. The role of bile acids in carcinogenesis. Cell Mol. Life Sci. 2022, 79, 243. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Toffanin, S.; Lachenmayer, A.; Villanueva, A.; Minguez, B.; Llovet, J.M. Molecular classification and novel targets in hepatocellular carcinoma: Recent advancements. Semin. Liver Dis. 2010, 30, 35–51. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Ito, H.; Wakai, K.; Nagata, C.; Mizoue, T.; Sasazuki, S.; Inoue, M.; et al. Obesity and liver cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2012, 42, 212–221. [Google Scholar] [CrossRef]
- Sohn, W.; Lee, H.W.; Lee, S.; Lim, J.H.; Lee, M.W.; Park, C.H.; Yoon, S.K. Obesity and the risk of primary liver cancer: A systematic review and meta-analysis. Clin. Mol. Hepatol. 2021, 27, 157–174. [Google Scholar] [CrossRef]
- He, Q.; Ding, Z.; Chen, T.; Wu, H.; Song, J.; Xiang, Z.; Yang, C.; Wang, S.; Xiong, B. PFDN2 promotes cell cycle progression via the hnRNPD-MYBL2 axis in gastric cancer. Front. Oncol. 2023, 13, 1164070. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Stopsack, K.H.; Wang, X.V.; Chen, Y.H.; Mazzu, Y.Z.; Burton, F.; Chakraborty, G.; Rajanala, S.H.; Hirani, R.; Nandakumar, S.; et al. Increased MYBL2 expression in aggressive hormone-sensitive prostate cancer. Mol. Oncol. 2022, 16, 3994–4010. [Google Scholar] [CrossRef]
- Zhao, J.Z.; Wang, W.; Liu, T.; Zhang, L.; Lin, D.Z.; Yao, J.Y.; Peng, X.; Jin, G.; Ma, T.T.; Gao, J.B.; et al. MYBL2 regulates de novo purine synthesis by transcriptionally activating IMPDH1 in hepatocellular carcinoma cells. BMC Cancer 2022, 22, 1290. [Google Scholar] [CrossRef]
- Wang, W.; Wu, S.; Shi, Y.; Miao, Y.; Luo, X.; Ji, M.; Yao, K.; He, J. c-MYB regulates cell growth and DNA damage repair through modulating MiR-143. FEBS Lett. 2015, 589, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.Y.; Zheng, D.F.; Lv, T.; Zhou, Y.J.; Yang, J.Y.; Jiang, L. Overexpression of transcription factor 3 drives hepatocarcinoma development by enhancing cell proliferation via activating Wnt signaling pathway. HBPD Int. 2022, 21, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Woody, S.; Sun, M.; Cui, W. Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver. Drug Metab. Rev. 2013, 45, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Xu, J.Y.; Shi, Z.; Englert, N.A.; Zhang, S.Y. Pregnane X receptor (PXR) deficiency improves high fat diet-induced obesity via induction of fibroblast growth factor 15 (FGF15) expression. Biochem. Pharmacol. 2017, 142, 194–203. [Google Scholar] [CrossRef]
- Haldar, S.M.; Jeyaraj, D.; Anand, P.; Zhu, H.; Lu, Y.; Prosdocimo, D.A.; Eapen, B.; Kawanami, D.; Okutsu, M.; Brotto, L.; et al. Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proc. Natl. Acad. Sci. USA 2012, 109, 6739–6744. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, R.; Jain, R.; Shi, H.; Zhang, L.; Zhou, G.; Sangwung, P.; Tugal, D.; Atkins, G.B.; Prosdocimo, D.A.; et al. Circadian control of bile acid synthesis by a KLF15-Fgf15 axis. Nat. Commun. 2015, 6, 7231. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, L.L.; Du, Y. Krüppel-like factor 15 in liver diseases: Insights into metabolic reprogramming. Front. Pharmacol. 2023, 14, 1115226. [Google Scholar] [CrossRef]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Sonke, G.S.; Hart, L.L.; Campone, M.; Erdkamp, F.; Janni, W.; Verma, S.; Villanueva, C.; Jakobsen, E.; Alba, E.; Wist, E.; et al. Ribociclib with letrozole vs letrozole alone in elderly patients with hormone receptor-positive, HER2-negative breast cancer in the randomized MONALEESA-2 trial. Breast Cancer Res. Treat. 2018, 167, 659–669. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef] [PubMed]
- Tut, T.G.; Lim, S.H.; Dissanayake, I.U.; Descallar, J.; Chua, W.; Ng, W.; de Souza, P.; Shin, J.S.; Lee, C.S. Upregulated Polo-Like Kinase 1 Expression Correlates with Inferior Survival Outcomes in Rectal Cancer. PLoS ONE 2015, 10, e0129313. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Lavia, P.; Guarguaglini, G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle 2006, 5, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Wang, Q.; Qiu, L.; Gong, X.; Chen, Z.; Zhang, H.; Ding, K.; Liu, Y.; Wei, J.; Lin, S.; et al. Combined Inhibition of UBE2C and PLK1 Reduce Cell Proliferation and Arrest Cell Cycle by Affecting ACLY in Pan-Cancer. Int. J. Mol. Sci. 2023, 24, 15658. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Eaton, J.E.; Silveira, M.G.; Pardi, D.S.; Sinakos, E.; Kowdley, K.V.; Luketic, V.A.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am. J. Gastroenterol. 2011, 106, 1638–1645. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Trauner, M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 432–450. [Google Scholar] [CrossRef]
- Lee, S.; Cho, Y.Y.; Cho, E.J.; Yu, S.J.; Lee, J.H.; Yoon, J.H.; Kim, Y.J. Synergistic effect of ursodeoxycholic acid on the antitumor activity of sorafenib in hepatocellular carcinoma cells via modulation of STAT3 and ERK. Int. J. Mol. Med. 2018, 42, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [PubMed]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, Y.; Gong, X.; Zhao, Z.; Zhang, Z.; Zhang, Q.; Huang, Y.; Xie, Q.; Liu, Y.; Wei, J.; Du, H. Establishment and Validation of Novel Prognostic Subtypes in Hepatocellular Carcinoma Based on Bile Acid Metabolism Gene Signatures Using Bulk and Single-Cell RNA-Seq Data. Int. J. Mol. Sci. 2024, 25, 919. https://doi.org/10.3390/ijms25020919
Qu Y, Gong X, Zhao Z, Zhang Z, Zhang Q, Huang Y, Xie Q, Liu Y, Wei J, Du H. Establishment and Validation of Novel Prognostic Subtypes in Hepatocellular Carcinoma Based on Bile Acid Metabolism Gene Signatures Using Bulk and Single-Cell RNA-Seq Data. International Journal of Molecular Sciences. 2024; 25(2):919. https://doi.org/10.3390/ijms25020919
Chicago/Turabian StyleQu, Yimo, Xiaocheng Gong, Ziyuan Zhao, Zimei Zhang, Qian Zhang, Yuting Huang, Qingsong Xie, Yunfei Liu, Jinfen Wei, and Hongli Du. 2024. "Establishment and Validation of Novel Prognostic Subtypes in Hepatocellular Carcinoma Based on Bile Acid Metabolism Gene Signatures Using Bulk and Single-Cell RNA-Seq Data" International Journal of Molecular Sciences 25, no. 2: 919. https://doi.org/10.3390/ijms25020919
APA StyleQu, Y., Gong, X., Zhao, Z., Zhang, Z., Zhang, Q., Huang, Y., Xie, Q., Liu, Y., Wei, J., & Du, H. (2024). Establishment and Validation of Novel Prognostic Subtypes in Hepatocellular Carcinoma Based on Bile Acid Metabolism Gene Signatures Using Bulk and Single-Cell RNA-Seq Data. International Journal of Molecular Sciences, 25(2), 919. https://doi.org/10.3390/ijms25020919