
Design of Two New Sulfur Derivatives of Perezone: In Silico Study Simulation Targeting PARP-1 and In Vitro Study Validation Using Cancer Cell Lines
 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
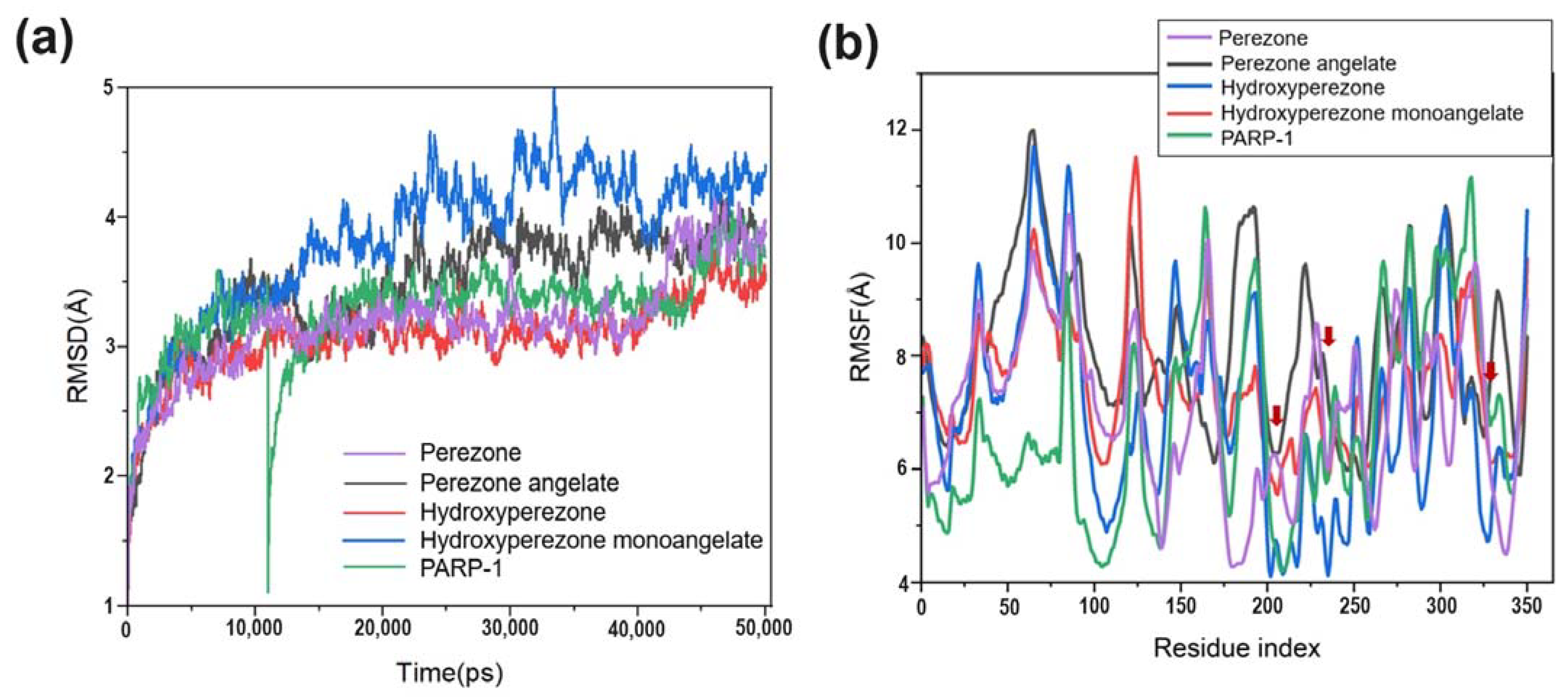
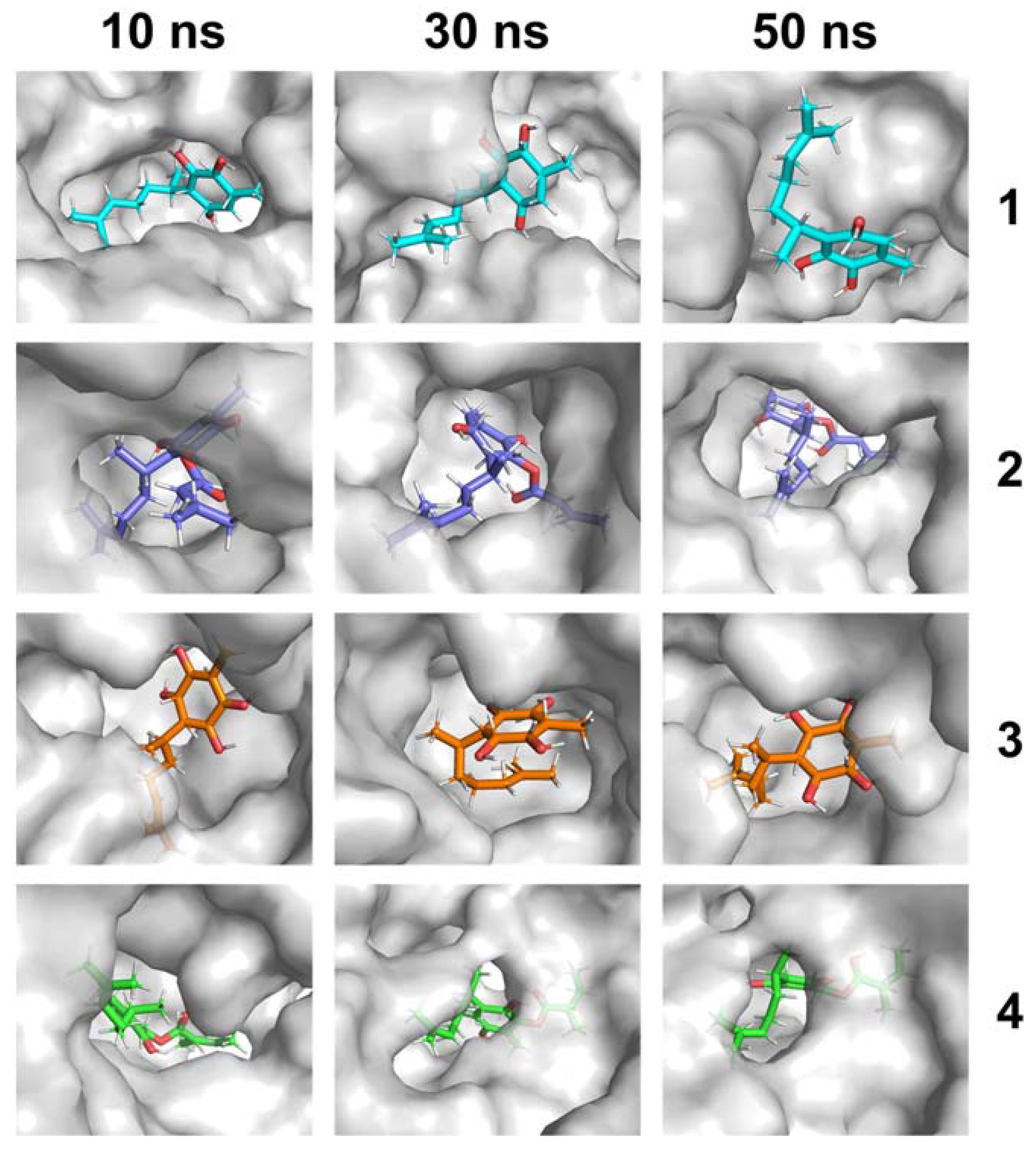
2.1. MD Simulations
2.2. Quantum Chemistry
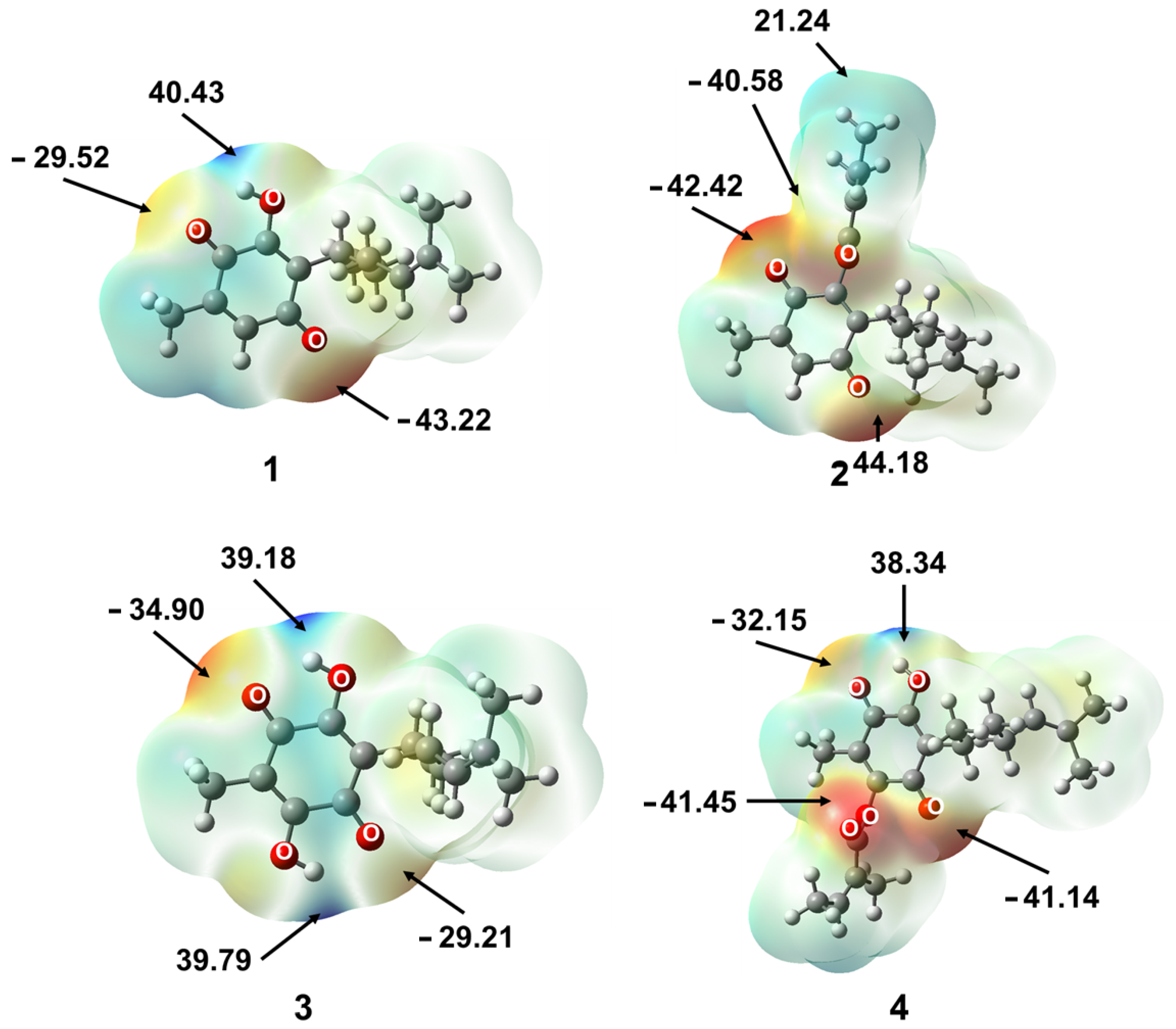
2.2.1. Molecular Electrostatic Potential (MPE)
2.2.2. Charge
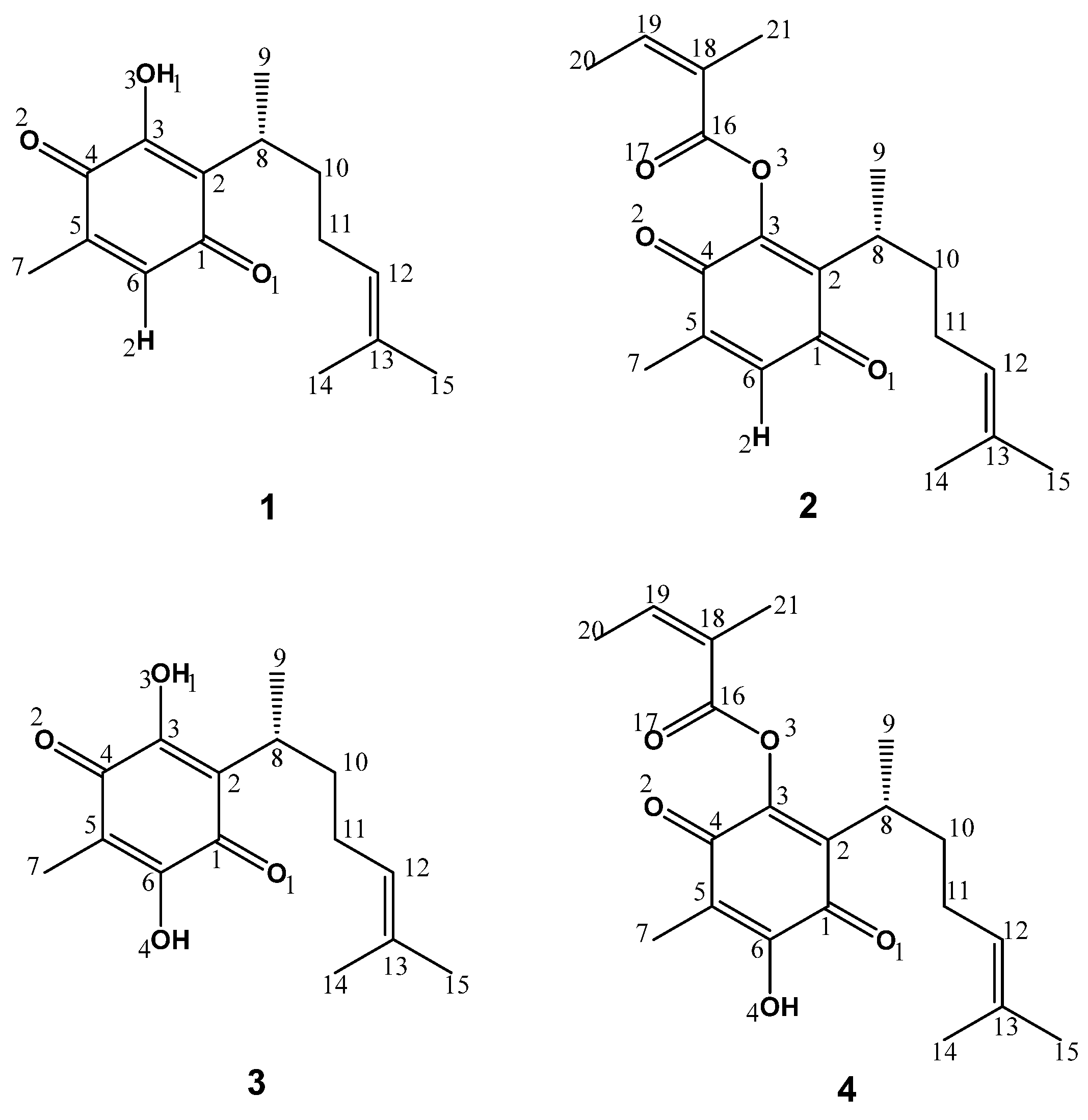
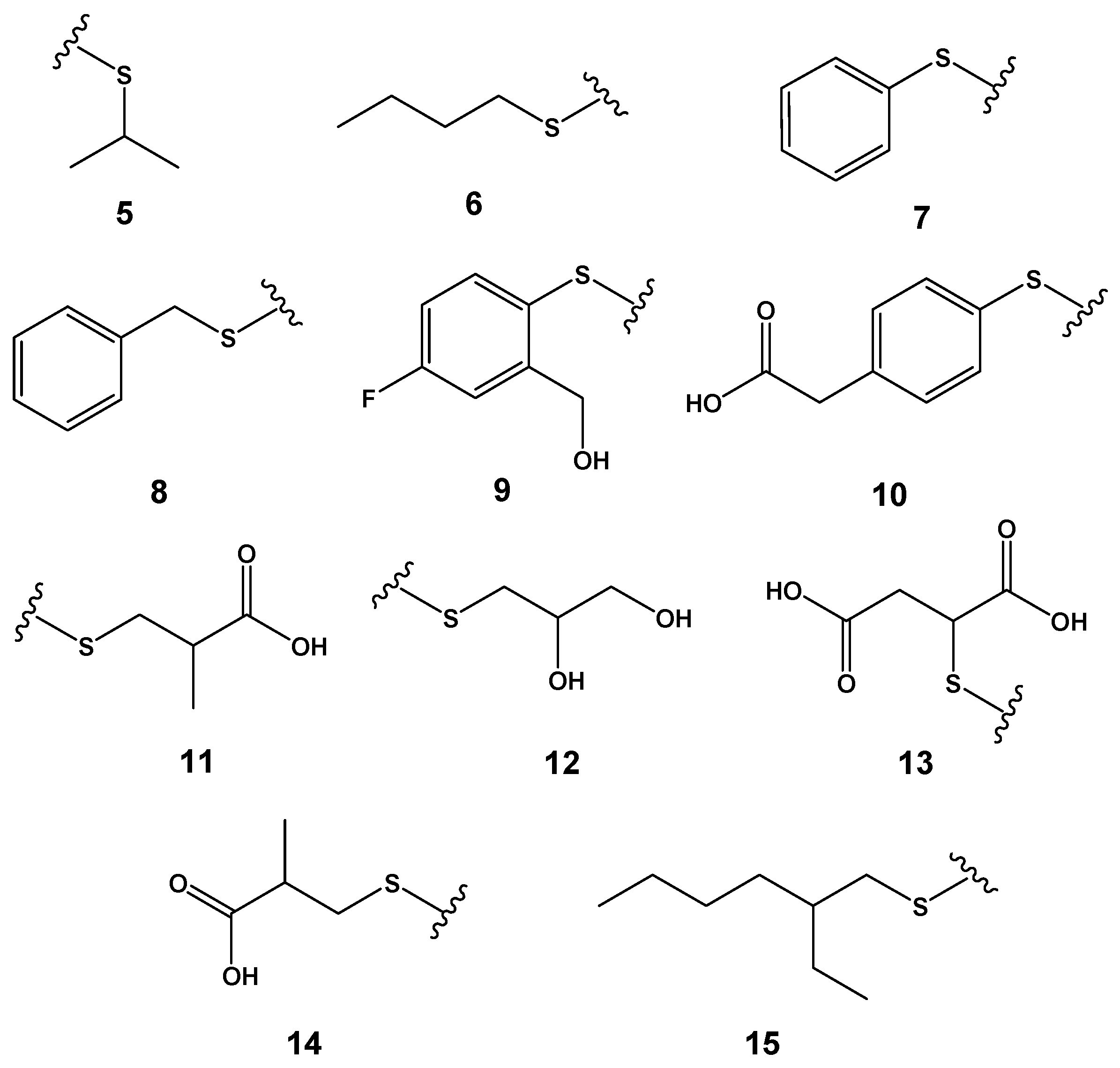
2.3. Design of Compounds
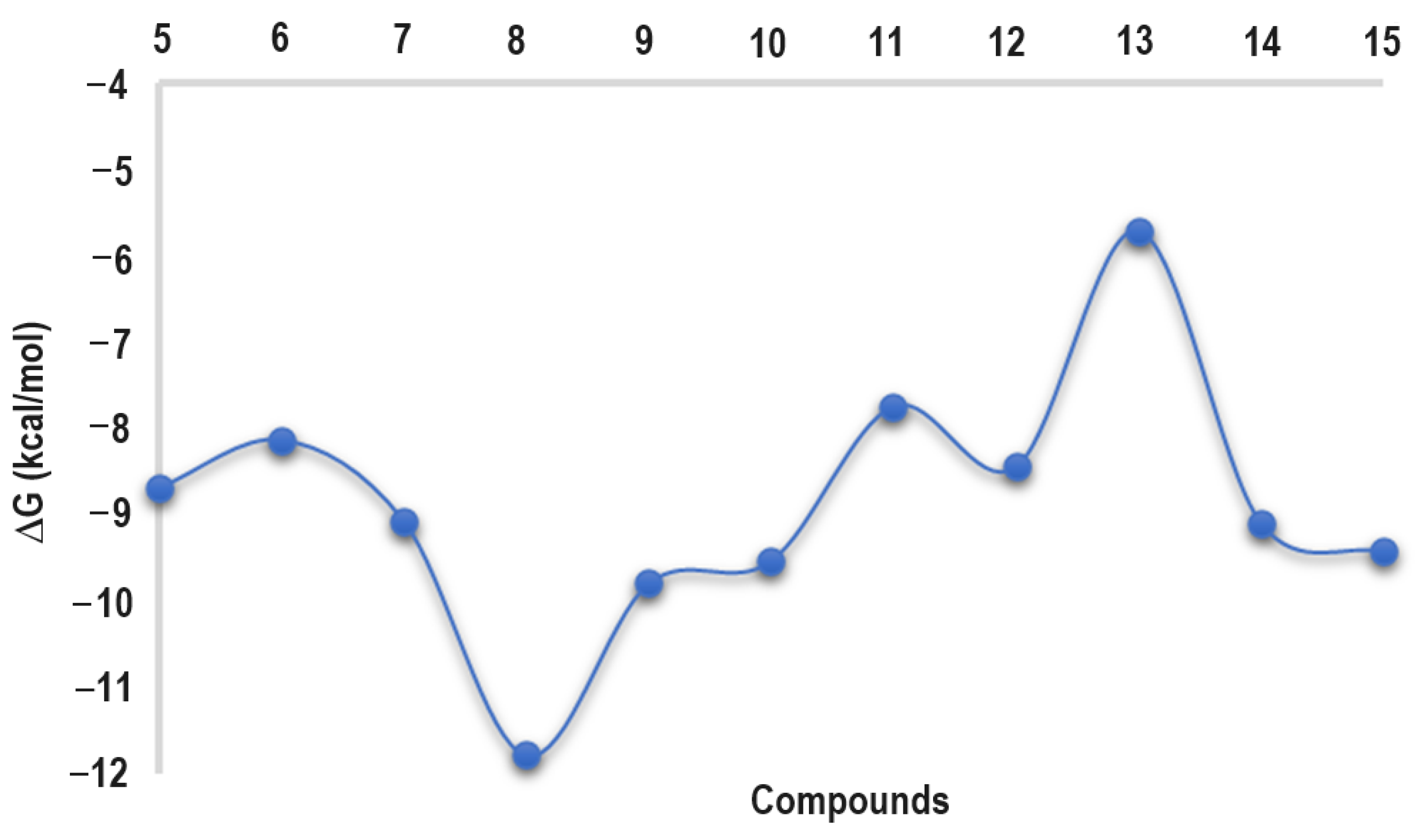
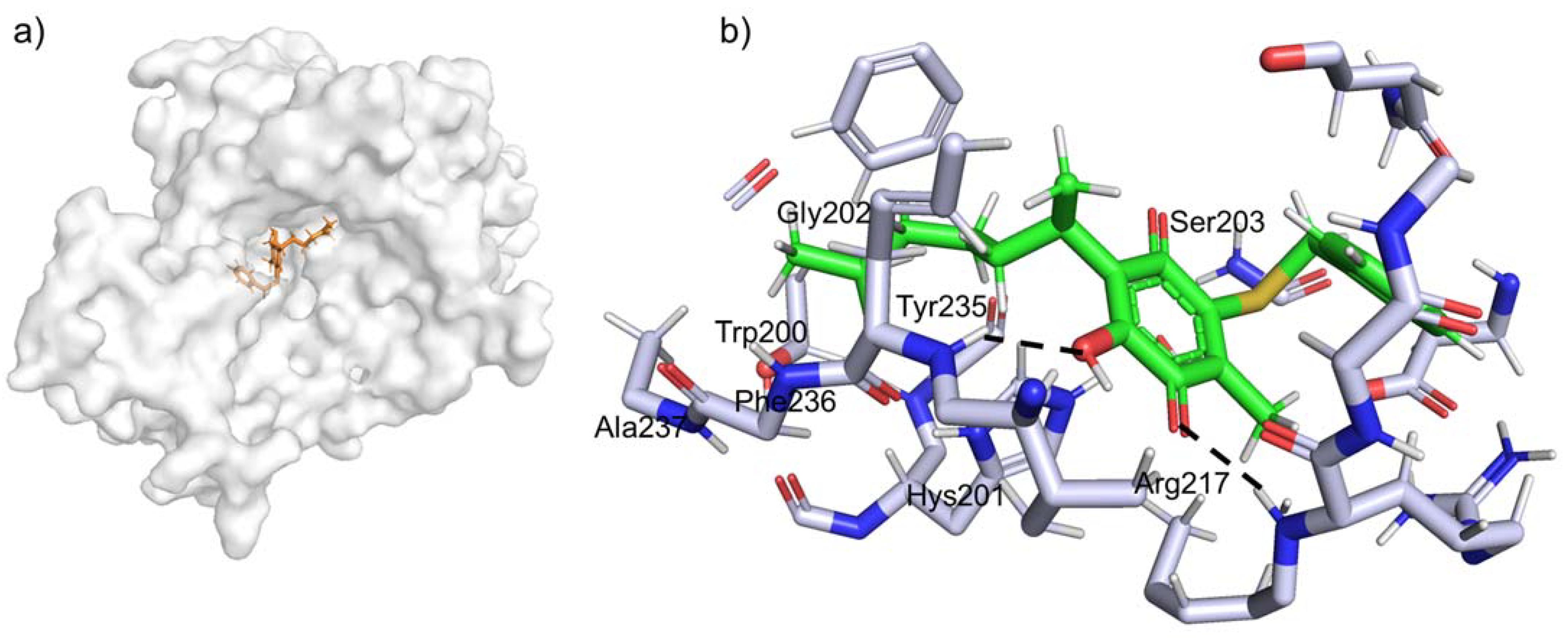
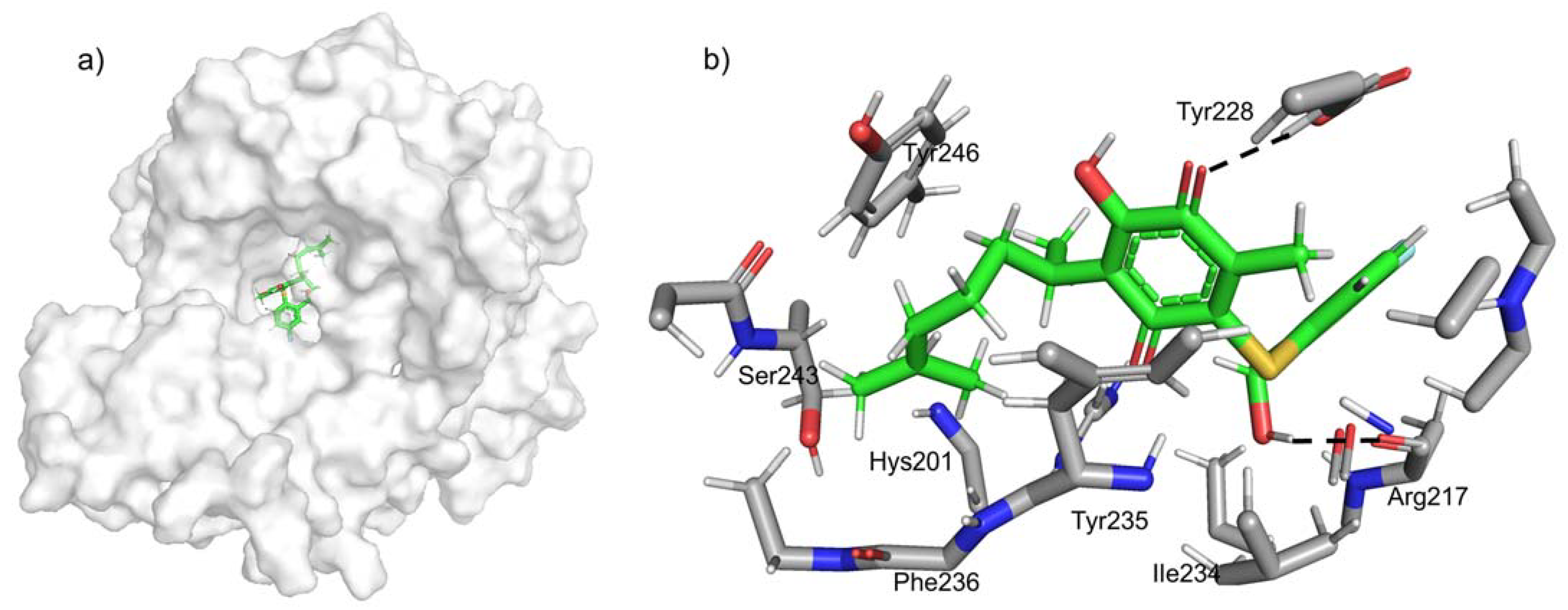
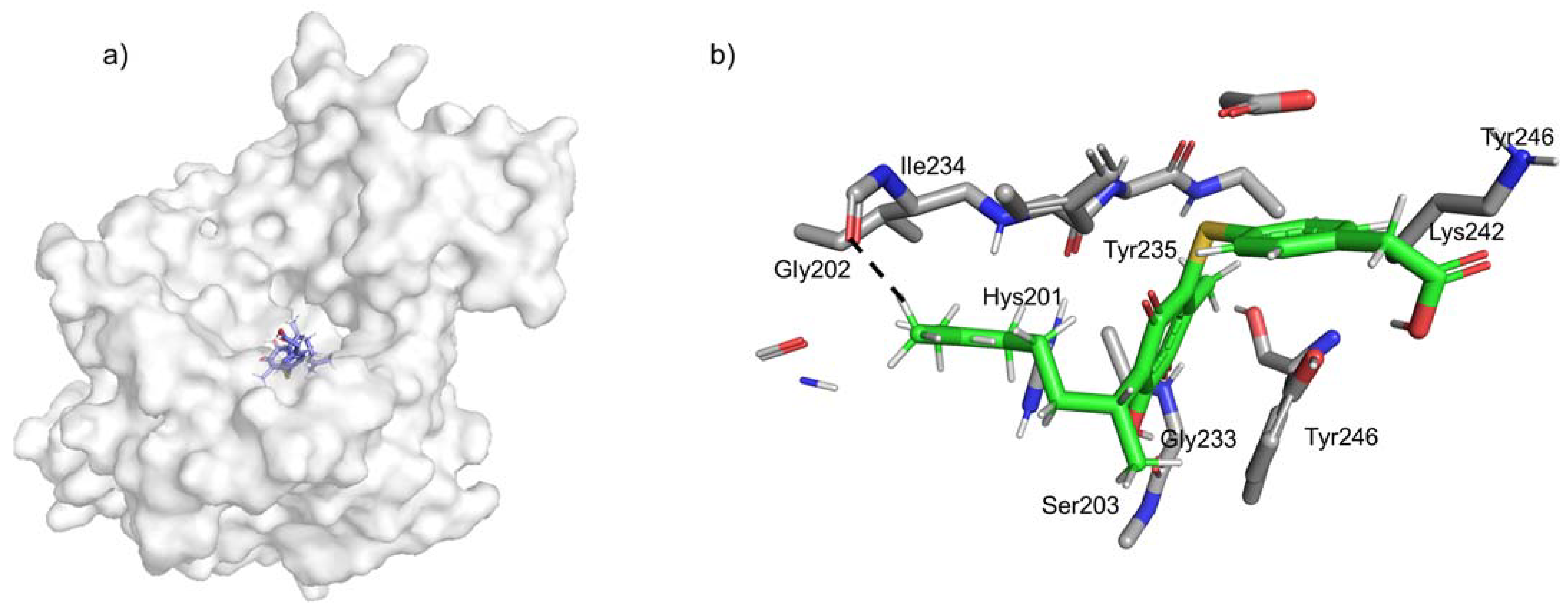
2.4. Docking Studies
2.5. Synthesis and Spectroscopic Attribution of 9 and 10
2.6. In Vitro Cytotoxic Assay
2.7. PARP-1 Inhibition Assay
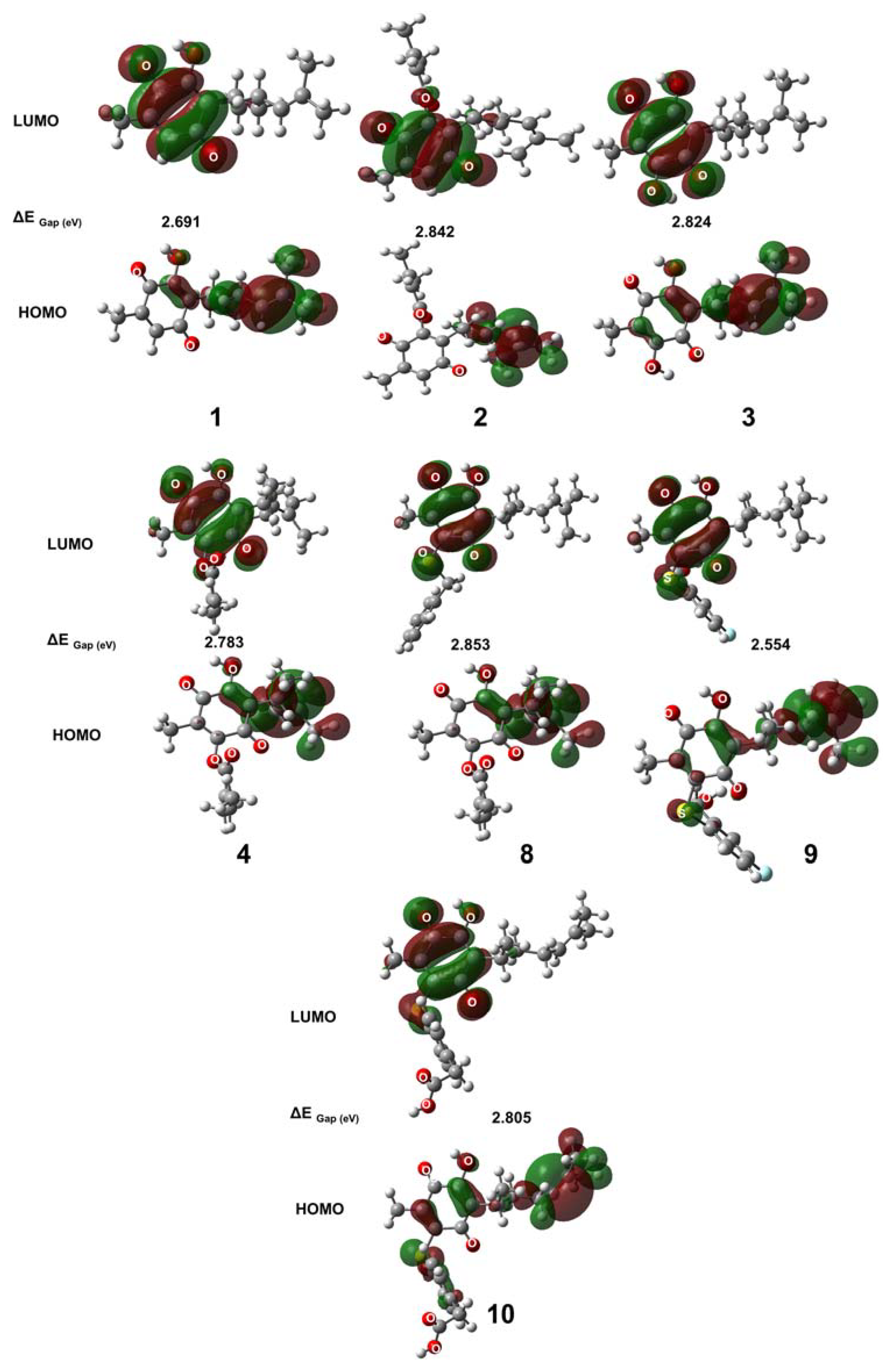
2.8. Quantum Properties and HOMO–LUMO Surface of 2–4 and 8–10
2.9. Reactivity Properties of 2–4 and 8–10
3. Materials and Methods
3.1. Molecular Dynamics Simulations of Four PARP-1 Inhibitors Related to Perezone Derivatives
3.2. Quantum Chemical Calculations
3.3. Design of Compounds
3.4. Synthesis of Compounds 8–10
3.4.1. General
3.4.2. Synthesis of 9 and 10
3.4.3. Spectroscopic Characterization of 9 and 10
3.5. PARP-1 Inhibition Assay
3.6. Cytotoxic Evaluation on Cancer Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in carcinomas and PARP1 inhibitors as antineoplastic drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Oliver, F.J.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Ménissier-de Murcia, J. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis: Lesson from an uncleavable mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef]
- Spiegel, J.O.; Van Houten, B.; Durrant, J.D. PARP1: Structural insights and pharmacological targets for inhibition. DNA Repair 2021, 103, 103125. [Google Scholar] [CrossRef]
- Río de la Loza, L. Discurso pronunciado por el catedrático de química médica de la Escuela de Medicina. In Escritos de Leopoldo Río de la Loza; Noriega, J.M., Ed.; Imprenta de Ignacio Escalante: Distrito Federal, Mexico, 1852; pp. 94–100. [Google Scholar]
- Lozada, M.C.; Soria-Arteche, O.; Ramírez-Apan, M.T.; Nieto-Camacho, A.; Enríquez, R.G.; Izquierdo, T.; Jiménez-Corona, A. Synthesis, cytotoxic and antioxidant evaluations of amino derivatives from perezone. Bioorg. Med. Chem. 2012, 20, 5077–5084. [Google Scholar] [CrossRef]
- Escobedo-González, R.G.; Bahena, L.; Arias Tellez, J.L.; Hinojosa Torres, J.; Miranda Ruvalcaba, R.; Aceves-Hernández, J.M. Characterization and comparison of perezone with some analogues. Experimental and theoretical study. J. Mol. Struct. 2015, 1097, 98–105. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Mendoza Sánchez, P.I.; Macias-Perez, M.E.; Rosales-Cruz, E.; Mera-Jiménez, E.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R. In vitro and computational studies showed that perezone inhibits PARP-1 and induces changes in the redox state of K562 cells. Arch. Biochem. Biophys. 2019, 671, 225–234. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Mendoza Sánchez, P.I.; Martínez, J.; Macías Pérez, M.E.; Rosales Cruz, E.; Żołek, T.; Maciejewska, D.; Miranda Ruvalcaba, R.; Mera Jiménez, E.; Nicolás-Vázquez, M.I. In Vitro and computational studies of perezone and perezone angelate as potential anti-glioblastoma multiforme agents. Molecules 2022, 27, 1565. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Mendoza Sánchez, P.I.; Macías Perez, M.E.; Rosales Cruz, E.; Mera Jiménez, E.; Aceves-Hernández, J.M.; Nicolás Vázquez, M.I.; Miranda Ruvalcaba, R. In vitro and computational studies of natural products related to perezone as anti-neoplastic agents. Biochimie 2020, 171, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Rosales Hernández, M.C.; Mendieta Wejebe, J.E.; Martínez Archundia, M.; Correa Basurto, J. Current tools and methods in Molecular Dynamics (MD) simulations for drug design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Zuckerman, D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 2009, 5, 23–48. [Google Scholar]
- Ayub, A.R.; Basharat, G.; Arshad, S.; Nazir, S.; Hamid, H.; Arshed, S.M.; Zahid, M.N.; Iqbal, J.; Ayub, K. A quantum mechanical investigation of nanocone oxide as a drug carrier for zidovudine: AIDS drug. J. Mol. Graph. Model. 2023, 125, 108611. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, C.; Rajesh, P.; Dhanalakshmi, E.; Gnanasambandan, T.; Priyadharshini, M. Structure conformational, molecular docking and computational investigation of methyl linoleate. Chem. Phys. Impact 2023, 7, 100300. [Google Scholar] [CrossRef]
- Brintha, T.; James, J.J.; Amalanathan, M.; Babu, P.J.; Mary, M.S.M. Experimental and theoretical investigation of structure activity relationship on L-lysine monohydrate for antioxidant efficacy. Chem. Phys. Impact 2023, 7, 100311. [Google Scholar] [CrossRef]
- Kubinyi, H. Hydrogen bonding: The last mystery in drug design? In Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies; Testa, B., van de Waterbeemd, H., Folkers, G., Guy, R., Eds.; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2001; pp. 513–524. [Google Scholar]
- Ramírez-Durán, L.A.; Rosales Hernández, M.C.; Hernández Rodríguez, M.; Elena Mendieta-Wejebe, J.; Trujillo-Ferrara, J.; Correa Basurto, J. Mapping myeloperoxidase to identify its promiscuity properties using docking and molecular dynamics simulations. Curr. Pharm. Des. 2013, 19, 2204–2215. [Google Scholar] [CrossRef]
- Martínez, J.; Velasco Bejarano, B.; Delgado, F.; Pozas, R.; Torres Domínguez, H.M.; Trujillo Ferrara, J.G.; Arroyo, G.A.; Miranda Ruvalcaba, R. Eco-contribution to the chemistry of perezone, a comparative study, using different modes of activation and solventless conditions. Nat. Prod. Commun. 2008, 3, 1465–1468. [Google Scholar] [CrossRef]
- Balachandran, V.; Parimala, K. Molecular structure, vibrational spectra, NBO analysis, first hyperpolarizability, and HOMO, LUMO studies of mesityl chloride by density functional methods. J. Mol. Struct. 2012, 1007, 136–145. [Google Scholar] [CrossRef]
- Tamer, Ö.; Avcı, D.; Atalay, Y. Synthesis, crystal structure, spectroscopic characterization and nonlinear optical properties of Co (II)-picolinate complex. Mater. Chem. Phys. 2015, 168, 138–146. [Google Scholar] [CrossRef]
- Abdou, A.; Omran, O.A.; Nafady, A.; Antipin, I.S. Structural, spectroscopic, FMOs, and non-linear optical properties exploration of three thiacaix (4) arenes derivatives. Arab. J. Chem. 2022, 15, 103656. [Google Scholar] [CrossRef]
- Russo, N.; Toscano, M.; Grand, A. Theoretical determination of electron affinity and ionization potential of DNA and RNA bases. J. Comput. Chem. 2000, 21, 1243–1250. [Google Scholar] [CrossRef]
- Mulliken, R.S. A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys. 1934, 2, 782–793. [Google Scholar] [CrossRef]
- Merza, M.M.; Hussein, F.M.; AL-ani, R.R. Physical properties and biological activity of methyldopa drug carrier cellulose derivatives. Theoretical study. Egypt. J. Chem. 2021, 64, 4081–4090. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Pérez, P. A comparative analysis of the electrophilicity of organic molecules between the computed IPs and EAs and the HOMO and LUMO energies. Chem. Phys. Lett. 2007, 438, 341–345. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- NAMD. Scalable Molecular Dynamics. 2016. Available online: http://www.ks.uiuc.edu/Research/namd/ (accessed on 17 October 2023).
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- William, H. VMD-visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Deppmeier, B.J.; Driessen, A.J.; Hehre, T.S.; Hehre, W.J.; Johnson, J.A.; Klunzinger, P.E.; Leonard, J.M.; Pham, I.; Pietro, W.J.; Jianguo, Y.; et al. Spartan 06 for Windows; Wavefunction Inc.: Irvine, CA, USA, 2006. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G. Density Fucntional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry, Proceedings of the Third International Congress of Quantum Chemistry, Kyoto, Japan, October 29–November 3 1979; Fukui, K., Pullman, B., Eds.; Springer: Dordrecht, The Netherlands, 1980; Volume 3, pp. 5–15. [Google Scholar]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Wiberg, K.B. Basis set effects on calculated geometries: 6-311++ G** vs. aug-cc-pVDZ. J. Comput. Chem. 2004, 25, 1342–1346. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.D., II; Keith, T.A.; Millam, J.M. GaussView 6.0.16 (64-bit Windows); Semichem, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Du, Q.S.; Huang, R.B.; Chou, K.C. Advances in visual representation of molecular potentials. Expert Opin. Drug Discov. 2010, 5, 513–527. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO Version 3; University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Dias, R.; de Azevedo, J.; Walter, F. Molecular docking algorithms. Curr. Drug Targets 2008, 9, 1040–1047. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System; Version 1.3r1; PyMOL Schödinger: New York, NY, USA, 2010. [Google Scholar]
- Escobedo-González, R.; Vázquez Cabañas, A.; Martínez González, A.; Mendoza Sánchez, P.; Saavedra-Leos, Z.; Cruz-Olivares, J.; Nava Serrano, J.; Martínez, J.; Miranda Ruvalcaba, R. Green approach Extraction of perezone from the roots of Acourtia platyphilla (A. Grey): A comparison of four activating modes and supercritical carbon dioxide. Molecules 2019, 24, 3035. [Google Scholar] [CrossRef]
- Escobedo-González, R.; Mendoza, P.; Nicolás-Vázquez, M.I.; Hernández-Rodríguez, M.; Martínez, J.; Miranda Ruvalcaba, R. A Timeline of Perezone, the First Isolated Secondary Metabolite in the New World, Covering the Period from 1852 to 2020. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Gibbons, S., Asakawa, Y., Liu, J.-K., Dirsch, V.M., Eds.; Springer: Berlin, Germany, 2021; Volume 116, pp. 67–133. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cell Lines | Compounds | |||
|---|---|---|---|---|
| 1 (Control) | 8 | 9 | 10 | |
| A-549 | 143.8 | >200 | >200 | >200 |
| MDA-MB-231 | 7.8 | >200 | 145.01 | >200 |
| HELA | 42.3 | 10.18 | 200 | >200 |
| MCF-7 | 53.2 | >200 | 83.17 | >200 |
| Normal cell lines | ||||
| Rat endothelial cells | >200 | >200 | >200 | >200 |
| VERO | 312.1 | 189 | 186 | 168 |
| Mix cells | 63.1 | >200 | 178.82 | >200 |
| Compounds | IC50 (µM) against PARP-1 Enzyme | |
|---|---|---|
| 8 | 5.205 | |
| 9 | 0.317 | |
| 10 | 0.320 | |
| Olaparib | 0.0063 | |
| Parameter | Compounds | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 8 | 9 | 10 | |
| ΔΕ Gap | 2.69 | 2.84 | 2.82 | 2.78 | 2.85 | 2.55 | 2.81 |
| E neutral | −809.5218 | −1078.9561 | −884.7844 | −1154.2104 | −1478.1656 | −1652.6668 | −1666.7941 |
| E positive | −809.2256 | −1078.6573 | −884.4907 | −1153.9186 | −1477.8862 | −1652.3797 | −1666.5141 |
| E negative | −809.5904 | −1079.0219 | −884.8511 | −1154.2802 | −1478.2356 | −1652.7455 | −1666.8686 |
| IP | 8.06 | 8.13 | 7.99 | 7.94 | 7.60 | 7.81 | 7.62 |
| EA | 1.87 | 1.79 | 1.82 | 1.90 | 1.90 | 2.14 | 2.03 |
| η | 3.10 | 3.17 | 3.09 | 3.02 | 2.85 | 2.84 | 2.80 |
| χ | 4.96 | 4.96 | 4.90 | 4.92 | 4.75 | 4.98 | 4.82 |
| μ | −4.96 | −4.96 | −4.90 | −4.92 | −4.75 | −4.98 | −4.82 |
| ω | 3.98 | 3.88 | 3.89 | 4.01 | 3.96 | 4.37 | 4.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubiales-Martínez, A.; Martínez, J.; Mera-Jiménez, E.; Pérez-Flores, J.; Téllez-Isaías, G.; Miranda Ruvalcaba, R.; Hernández-Rodríguez, M.; Mancilla Percino, T.; Macías Pérez, M.E.; Nicolás-Vázquez, M.I. Design of Two New Sulfur Derivatives of Perezone: In Silico Study Simulation Targeting PARP-1 and In Vitro Study Validation Using Cancer Cell Lines. Int. J. Mol. Sci. 2024, 25, 868. https://doi.org/10.3390/ijms25020868
Rubiales-Martínez A, Martínez J, Mera-Jiménez E, Pérez-Flores J, Téllez-Isaías G, Miranda Ruvalcaba R, Hernández-Rodríguez M, Mancilla Percino T, Macías Pérez ME, Nicolás-Vázquez MI. Design of Two New Sulfur Derivatives of Perezone: In Silico Study Simulation Targeting PARP-1 and In Vitro Study Validation Using Cancer Cell Lines. International Journal of Molecular Sciences. 2024; 25(2):868. https://doi.org/10.3390/ijms25020868
Chicago/Turabian StyleRubiales-Martínez, Alejandro, Joel Martínez, Elvia Mera-Jiménez, Javier Pérez-Flores, Guillermo Téllez-Isaías, René Miranda Ruvalcaba, Maricarmen Hernández-Rodríguez, Teresa Mancilla Percino, Martha Edith Macías Pérez, and María Inés Nicolás-Vázquez. 2024. "Design of Two New Sulfur Derivatives of Perezone: In Silico Study Simulation Targeting PARP-1 and In Vitro Study Validation Using Cancer Cell Lines" International Journal of Molecular Sciences 25, no. 2: 868. https://doi.org/10.3390/ijms25020868
APA StyleRubiales-Martínez, A., Martínez, J., Mera-Jiménez, E., Pérez-Flores, J., Téllez-Isaías, G., Miranda Ruvalcaba, R., Hernández-Rodríguez, M., Mancilla Percino, T., Macías Pérez, M. E., & Nicolás-Vázquez, M. I. (2024). Design of Two New Sulfur Derivatives of Perezone: In Silico Study Simulation Targeting PARP-1 and In Vitro Study Validation Using Cancer Cell Lines. International Journal of Molecular Sciences, 25(2), 868. https://doi.org/10.3390/ijms25020868