
Modulation of Tau Pathology in Alzheimer’s Disease by Dietary Bioactive Compounds


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tauopathy in AD
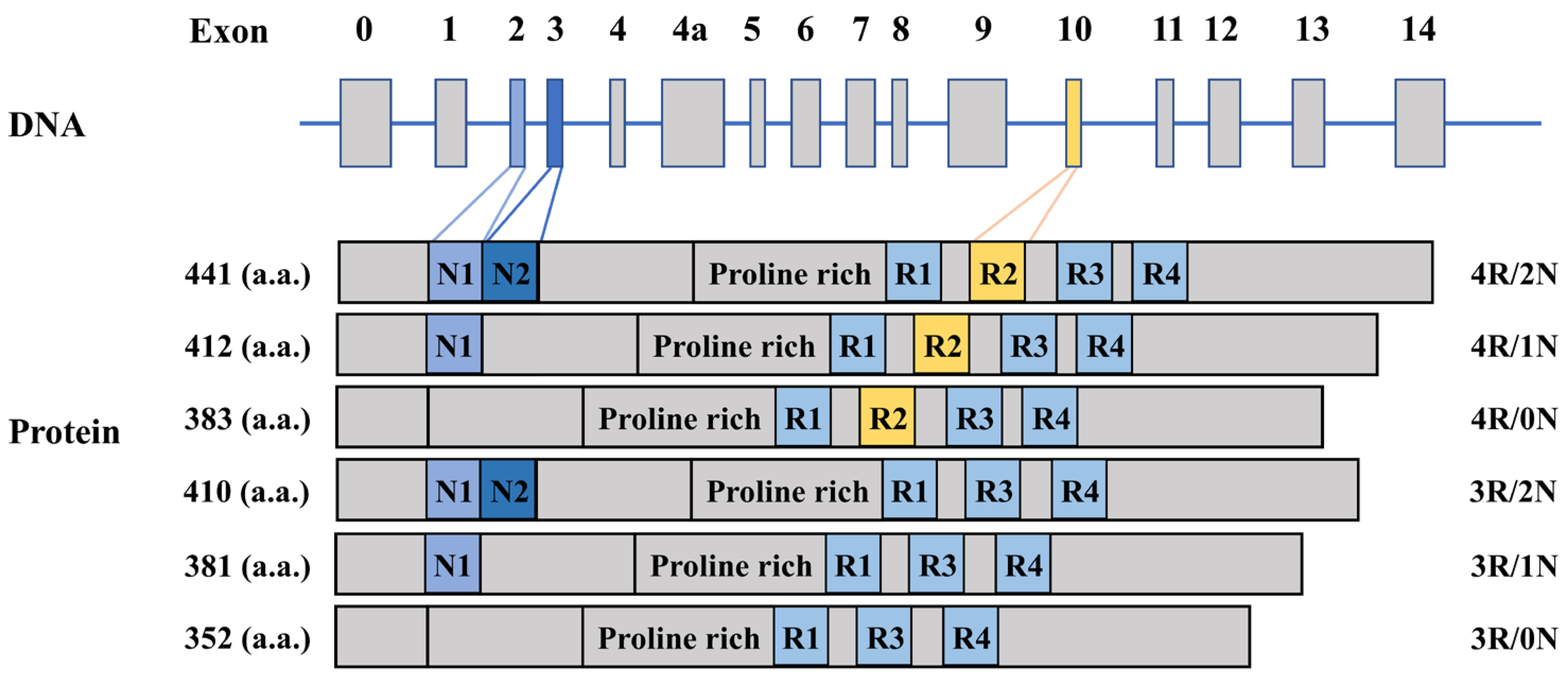
2.1. The Gene and Function of Tau Protein
2.2. Post-Translational Modification of Tau Protein in Physiological and Pathological Conditions
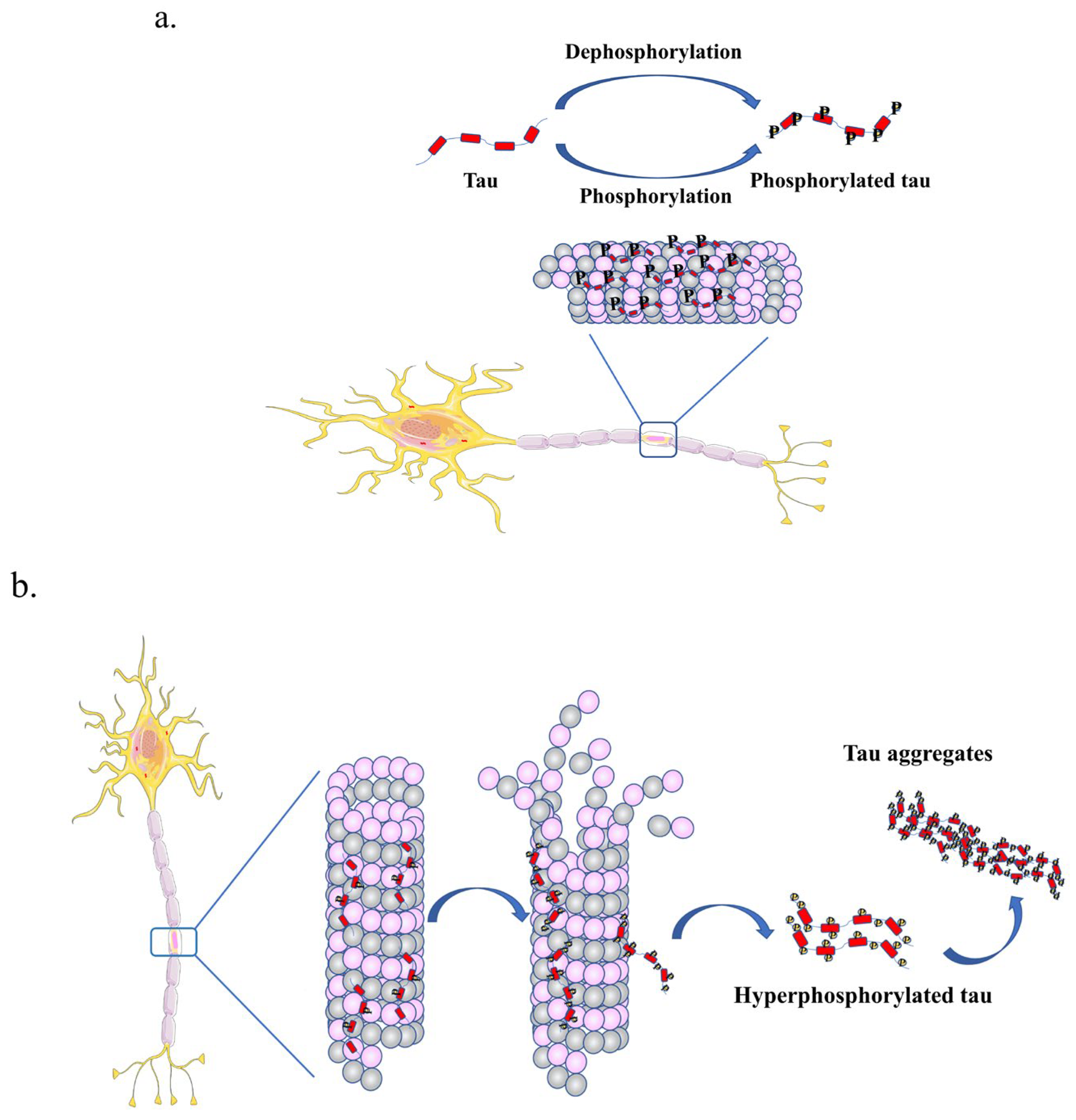
2.2.1. Phosphorylation of Tau
2.2.2. SUMOylation of Tau
2.2.3. Acetylation of Tau
2.3. Tau Aggregation in Tauopathy
3. Clearance of Misfolded Tau by Protein Degradation System
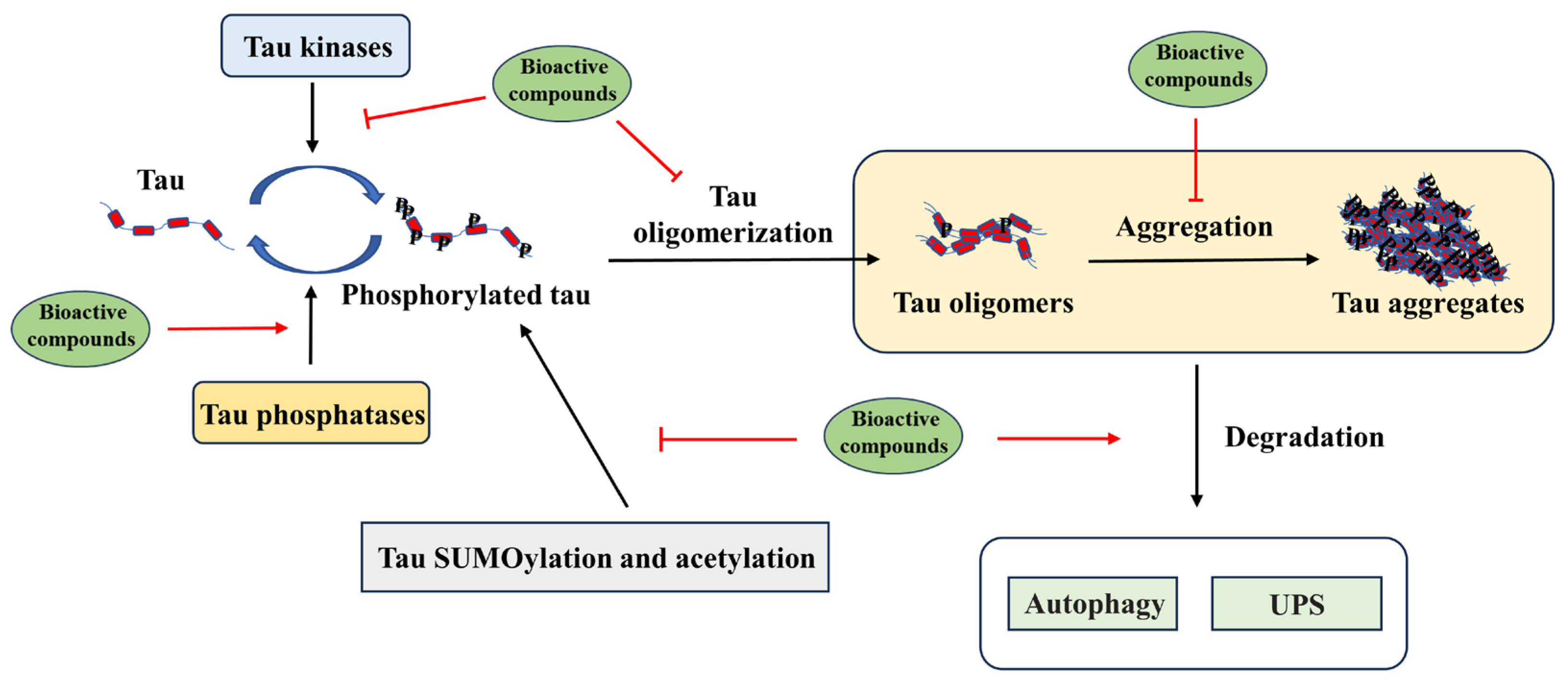
4. Modulation of Tau Pathology by Dietary Bioactive Compounds
4.1. Targeting Tau Post-Translational Modification by Bioactive Compounds
4.2. Targeting Tau Aggregation by Dietary Bioactive Compounds
4.3. Targeting Tau Degradation by Dietary Bioactive Compounds
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.X.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef] [PubMed]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.; Kouri, N.; Murray, M.; Josephs, K. Neuropathology of Frontotemporal Lobar Degeneration-Tau (FTLD-Tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef]
- Wray, S.; Saxton, M.; Anderton, B.H.; Hanger, D.P. Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J. Neurochem. 2008, 105, 2343–2352. [Google Scholar] [CrossRef]
- Robinson, J.L.; Yan, N.; Caswell, C.; Xie, S.X.; Suh, E.; Van Deerlin, V.M.; Gibbons, G.; Irwin, D.J.; Grossman, M.; Lee, E.B.; et al. Primary Tau Pathology, Not Copathology, Correlates with Clinical Symptoms in PSP and CBD. J. Neuropathol. Exp. Neurol. 2020, 79, 296–304. [Google Scholar] [CrossRef]
- Zhang, R.F.; Zeng, M.; Zhang, X.L.; Zheng, Y.J.; Lv, N.; Wang, L.M.; Gan, J.L.; Li, Y.W.; Jiang, X.J.; Yang, L. Therapeutic Candidates for Alzheimer’s Disease: Saponins. Int. J. Mol. Sci. 2023, 24, 10505. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I.; Smith, A.J.; George, L.; Tung, Y.C.; Zaidi, T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 5646–5650. [Google Scholar] [CrossRef]
- Haroutunian, V.; Schnaider-Beeri, M.; Schmeidler, J.; Wysocki, M.; Purohit, D.P.; Perl, D.P.; Libow, L.S.; Lesser, G.T.; Maroukian, M.; Grossman, H.T. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch. Neurol. 2008, 65, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Rayman, J.B. Focusing on oligomeric tau as a therapeutic target in Alzheimer’s disease and other tauopathies. Expert Opin. Ther. Targets 2023, 27, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Khanna, M.R.; Kovalevich, J.; Lee, V.M.Y.; Trojanowski, J.Q.; Brunden, K.R. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimer’s Dement. 2016, 12, 1051–1065. [Google Scholar] [CrossRef]
- Mullard, A. Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat. Rev. Drug Discov. 2021, 20, 3–5. [Google Scholar] [CrossRef]
- Congdon, E.E.; Ji, C.Y.; Tetlow, A.M.; Jiang, Y.X.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease: Current status and future directions. Nat. Rev. Neurol. 2023, 19, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, R.; Ohishi, T.; Koyama, Y.; Pervin, M.; Nakamura, Y.; Isemura, M. Beneficial Effects of Epigallocatechin-3-O-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases. Molecules 2021, 26, 415. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Yamada, M. Curcumin and Alzheimer’s Disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Calcul, L.; Zhang, B.; Jinwal, U.K.; Dickey, C.A.; Baker, B.J. Natural products as a rich source of tau-targeting drugs for Alzheimer’s disease. Future Med. Chem. 2012, 4, 1751–1761. [Google Scholar] [CrossRef]
- Grodzicki, W.; Dziendzikowska, K. The Role of Selected Bioactive Compounds in the Prevention of Alzheimer’s Disease. Antioxidants 2020, 9, 229. [Google Scholar] [CrossRef]
- Veurink, G.; Perry, G.; Singh, S.K. Role of antioxidants and a nutrient rich diet in Alzheimer’s disease. Open. Biol. 2020, 10, 16. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 1986, 1, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Bombieri, C.; Valenti, M.T.; Romanelli, M.G. Tau Isoforms: Gaining Insight into MAPT Alternative Splicing. Int. J. Mol. Sci. 2022, 23, 15383. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-Specific Functional Impairments in Distinct Tau Isoforms of Hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. Embo J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Funakoshi, T.; SatoHarada, R.; Kanai, Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 1996, 132, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; Whitcomb, D.J.; Cho, K. Physiological and Pathophysiological Implications of Synaptic Tau. Neuroscientist 2017, 23, 137–151. [Google Scholar] [CrossRef]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- Best, R.L.; LaPointe, N.E.; Liang, J.H.; Ruan, K.; Shade, M.F.; Wilson, L.; Feinstein, S.C. Tau isoform-specific stabilization of intermediate states during microtubule assembly and disassembly. J. Biol. Chem. 2019, 294, 12265–12280. [Google Scholar] [CrossRef]
- Christensen, K.R.; Combs, B.; Richards, C.; Grabinski, T.; Alhadidy, M.M.; Kanaan, N.M. Phosphomimetics at Ser199/Ser202/Thr205 in Tau Impairs Axonal Transport in Rat Hippocampal Neurons. Mol. Neurobiol. 2023, 16, 3423–3438. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.Q.; Han, Y.; Li, P.; Su, Z.D.; Huang, Y.Q. The Role of Post-Translational Modifications on the Structure and Function of Tau Protein. J. Mol. Neurosci. 2022, 72, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Acosta, D.M.; Mancinelli, C.; Bracken, C.; Eliezer, D. Post-translational modifications within tau paired helical filament nucleating motifs perturb microtubule interactions and oligomer formation. J. Biol. Chem. 2022, 298, 101442. [Google Scholar] [CrossRef] [PubMed]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef] [PubMed]
- Biernat, J.; Mandelkow, E.M. The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol. Biol. Cell 1999, 10, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Luna-Viramontes, N.I.; Campa-Córdoba, B.B.; Ontiveros-Torres, M.A.; Harrington, C.R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; de la Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S.; et al. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 247. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Terro, F. Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef]
- Wang, Y.X.; Yang, R.Y.; Gu, J.L.; Yin, X.M.; Jin, N.N.; Xie, S.T.; Wang, Y.F.; Chang, H.H.; Qian, W.; Shi, J.H.; et al. Cross talk between PI3K-AKT-GSK-3β and PP2A pathways determines tau hyperphosphorylation. Neurobiol. Aging 2015, 36, 188–200. [Google Scholar] [CrossRef]
- Avila, J. Tau kinases and phosphatases. J. Cell. Mol. Med. 2008, 12, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.-R.; Zhu, L.-Q.; Wang, S.-H.; Liu, X.-A.; Tian, Q.; Zhang, Q.; Wang, Q.; Wang, J.-Z. 17β-estradiol attenuates glycogen synthase kinase-3β activation and tau hyperphosphorylation in Akt-independent manner. J. Neural Transm. 2008, 115, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Johnson, G.V.W. Glycogen synthase kinase 3 beta phosphorylates tau at both primed and unprimed sites—Differential impact on microtubule binding. J. Biol. Chem. 2003, 278, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Wu, Q.L.; Smith, A.; Grundke-Iqbal, I.; Iqbal, K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998, 436, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Johnson, G.V.W. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J. Neurochem. 2004, 88, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef]
- Marcus, J.N.; Schachter, J. Targeting post-translational modifications on tau as a therapeutic strategy for Alzheimer’s disease. J. Neurogenet. 2011, 25, 127–133. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.B.; Gao, J.; Wang, J.M. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2023, 10, 458–467. [Google Scholar] [CrossRef]
- Tochio, N.; Murata, T.; Utsunomiya-Tate, N. Effect of site-specific amino acid D-isomerization on β-sheet transition and fibril formation profiles of Tau microtubule-binding repeat peptides. Biochem. Biophys. Res. Commun. 2019, 508, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Le, L.; Lee, J.; Im, D.; Park, S.; Hwang, K.D.; Lee, J.H.; Jiang, Y.; Lee, Y.S.; Suh, Y.H.; Kim, H.I.; et al. Self-Aggregating Tau Fragments Recapitulate Pathologic Phenotypes and Neurotoxicity of Alzheimer’s Disease in Mice. Adv. Sci. 2023, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; GrundkeIqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Drisaldi, B.; Colnaghi, L.; Fioriti, L.; Rao, N.; Myers, C.; Snyder, A.M.; Metzger, D.J.; Tarasoff, J.; Konstantinov, E.; Fraser, P.E.; et al. SUMOylation Is an Inhibitory Constraint that Regulates the Prion-like Aggregation and Activity of CPEB3. Cell Rep. 2015, 11, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Werner, A.; Takahashi-Fujigasaki, J.; Daret, A.; Fujigasaki, H.; Takada, K.; Duyckaerts, C.; Brice, A.; Dejean, A.; Sittler, A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 2010, 19, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef]
- Qi, Q.; Liu, X.; Brat, D.J.; Ye, K. Merlin sumoylation is required for its tumor suppressor activity. Oncogene 2014, 33, 4893–4903. [Google Scholar] [CrossRef]
- Truong, K.; Lee, T.D.; Li, B.Z.; Chen, Y. Sumoylation of SAE2 C Terminus Regulates SAE Nuclear Localization. J. Biol. Chem. 2012, 287, 42611–42619. [Google Scholar] [CrossRef]
- Jaafari, N.; Konopacki, F.A.; Owen, T.F.; Kantamneni, S.; Rubin, P.; Craig, T.J.; Wilkinson, K.A.; Henley, J.M. SUMOylation Is Required for Glycine-Induced Increases in AMPA Receptor Surface Expression (ChemLTP) in Hippocampal Neurons. PLoS ONE 2013, 8, e52345. [Google Scholar] [CrossRef]
- Luo, H.B.; Xia, Y.Y.; Shu, X.J.; Liu, Z.C.; Feng, Y.; Liu, X.H.; Yu, G.; Yin, G.; Xiong, Y.S.; Zeng, K.; et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Sakurai, M.; Matsuzaki, S.; Arancio, O.; Fraser, P. SUMO and Alzheimer’s Disease. Neuromol. Med. 2013, 15, 720–736. [Google Scholar] [CrossRef]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Feinstein, S.C. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 1994, 124, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Guan, L.L. Unraveling the Influence of K280 Acetylation on the Conformational Features of Tau Core Fragment: A Molecular Dynamics Simulation Study. Front. Mol. Biosci. 2021, 8, 11. [Google Scholar] [CrossRef]
- Wang, Y.P.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.Q.; Zhou, Y.G.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef]
- Xia, Y.X.; Bell, B.M.; Giasson, B.I. Tau K321/K353 pseudoacetylation within KXGS motifs regulates tau-microtubule interactions and inhibits aggregation. Sci. Rep. 2021, 11, 9. [Google Scholar] [CrossRef]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef]
- Carlonnagno, Y.; Chung, D.E.C.; Yue, M.; Castanedes-Casey, M.; Madden, B.J.; Dunmore, J.; Tong, J.M.; DeTure, M.; Dickson, D.W.; Petrucelli, L.; et al. An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem. 2017, 292, 15277–15286. [Google Scholar] [CrossRef] [PubMed]
- Gorsky, M.K.; Burnouf, S.; Dols, J.; Mandelkow, E.; Partridge, L. Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci. Rep. 2016, 6, 22685. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, I.; Varghese, M.; Ksiezak-Reding, H.; Dzhun, A.; Wang, J.; Pasinetti, G.M. Paired Helical Filaments from Alzheimer Disease Brain Induce Intracellular Accumulation of Tau Protein in Aggresomes. J. Biol. Chem. 2012, 287, 20522–20533. [Google Scholar] [CrossRef] [PubMed]
- Tarutani, A.; Adachi, T.; Akatsu, H.; Hashizume, Y.; Hasegawa, K.; Saito, Y.; Robinson, A.C.; Mann, D.M.A.; Yoshida, M.; Murayama, S.; et al. Ultrastructural and biochemical classification of pathogenic tau, α-synuclein and TDP-43. Acta Neuropathol. 2022, 143, 613–640. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Paired Helical Filaments in Electron Microscopy of Alzheimer’s Disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Ferrer, I.; Pérez, M.; Zabala, J.C.; del Rio, J.A.; Avila, J. Tau Aggregation. Neuroscience 2023, 518, 6. [Google Scholar] [CrossRef]
- Wang, D.; Huang, X.L.; Yan, L.; Zhou, L.Q.; Yan, C.; Wu, J.H.; Su, Z.D.; Huang, Y.Q. The Structure Biology of Tau and Clue for Aggregation Inhibitor Design. Protein J. 2021, 40, 656–668. [Google Scholar] [CrossRef]
- Aillaud, I.; Funke, S.A. Tau Aggregation Inhibiting Peptides as Potential Therapeutics for Alzheimer Disease. Cell. Mol. Neurobiol. 2023, 43, 951–961. [Google Scholar] [CrossRef]
- Shah, S.J.A.; Zhang, Q.; Guo, J.; Liu, H.; Liu, H.; Vill-Freixa, J. Identification of Aggregation Mechanism of Acetylated PHF6*and PHF6 Tau Peptides Based on Molecular Dynamics Simulations and Markov State Modeling. ACS Chem. Neurosci. 2023, 14, 3959–3971. [Google Scholar] [CrossRef]
- Chang, E.; Kim, S.; Schafer, K.N.; Kuret, J. Pseudophosphorylation of tau protein directly modulates its aggregation kinetics. BBA-Proteins Proteom. 2011, 1814, 388–395. [Google Scholar] [CrossRef]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M.Y. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.L.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Despres, C.; Byrne, C.; Qi, H.; Cantrelle, F.X.; Huvent, I.; Chambraud, B.; Baulieu, E.E.; Jacquot, Y.; Landrieu, I.; Lippens, G.; et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 9080–9085. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.; Combs, B.; Abdelmesih, B.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Analysis of isoform-specific tau aggregates suggests a common toxic mechanism involving similar pathological conformations and axonal transport inhibition. Neurobiol. Aging 2016, 47, 113–126. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.Y.; Andreadis, A.; Fu, Y.F.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The Amino Terminus of Tau Inhibits Kinesin-Dependent Axonal Transport: Implications for Filament Toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef]
- Hintermayer, M.A.; Volkening, K.; Moszczynski, A.J.; Donison, N.; Strong, M.J. Tau protein phosphorylation at Thr175 initiates fibril formation via accessibility of the N-terminal phosphatase-activating domain. J. Neurochem. 2020, 155, 313–326. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Sugino, E.; Nishiura, C.; Minoura, K.; In, Y.; Sumida, M.; Taniguchi, T.; Tomoo, K.; Ishida, T. Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochem. Biophys. Res. Commun. 2009, 385, 236–240. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Niewiadomski, W.; Steczkowska, M.; Gasiorowska, A. Tau Oligomers Neurotoxicity. Life 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Kayed, R. Formation and propagation of tau oligomeric seeds. Front. Neurol. 2013, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. The prion-like transmission of tau oligomers via exosomes. Front. Aging Neurosci. 2022, 14, 974414. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef]
- Samimi, N.; Asada, A.; Ando, K. Tau Abnormalities and Autophagic Defects in Neurodegenerative Disorders; A Feed-forward Cycle. Galen Med. J. 2020, 9, 8. [Google Scholar] [CrossRef]
- Weng, F.L.; He, L. Disrupted ubiquitin proteasome system underlying tau accumulation in Alzheimer’s disease. Neurobiol. Aging 2021, 99, 79–85. [Google Scholar] [CrossRef]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Molec. Neurosci. 2020, 13, 586731. [Google Scholar] [CrossRef]
- Yang, L.; Guo, W.; Zhang, S.; Wang, G. Ubiquitination-proteasome system: A new player in the pathogenesis of psoriasis and clinical implications. J. Dermatol. Sci. 2018, 89, 219–225. [Google Scholar] [CrossRef]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Layfield, R.; Serpell, L.; Narain, Y.; Goedert, M.; Spillantini, M.G. Proteasomal degradation of tau protein. J. Neurochem. 2002, 83, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.E.; Djakovic, S.N.; Salehi, A.; Wilson, S.M.; Masliah, E.; Patrick, G.N. Regulation of Synaptic Structure by Ubiquitin C-Terminal Hydrolase L1. J. Neurosci. 2009, 29, 7857–7868. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef]
- Xie, M.; Han, Y.; Yu, Q.T.; Wang, X.; Wang, S.H.; Liao, X.M. UCH-L1 Inhibition Decreases the Microtubule-Binding Function of Tau Protein. J. Alzheimer’s Dis. 2016, 49, 353–363. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Cell biology—Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci. Ther. 2020, 26, 155–166. [Google Scholar] [CrossRef]
- Feng, Q.; Luo, Y.; Zhang, X.N.; Yang, X.F.; Hong, X.Y.; Sun, D.S.; Li, X.C.; Hu, Y.; Li, X.G.; Zhang, J.F.; et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: A vicious cycle in Alzheimer neurodegeneration. Autophagy 2020, 16, 641–658. [Google Scholar] [CrossRef]
- Piras, A.; Collin, L.; Gruninger, F.; Graff, C.; Ronnback, A. Autophagic and lysosomal defects in human tauopathies: Analysis of postmortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.H.; Lin, W.L.; Isidoro, C.; DeTure, M.; Ko, L.W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130. [Google Scholar] [CrossRef]
- Olmos, Y. The ESCRT Machinery: Remodeling, Repairing, and Sealing Membranes. Membranes 2022, 12, 633. [Google Scholar] [CrossRef] [PubMed]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Furlan, V.; Konc, J.; Bren, U. Inverse Molecular Docking as a Novel Approach to Study Anticarcinogenic and Anti-Neuroinflammatory Effects of Curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Ooizumi, T.; Kawakami, F.; Lwin, T.T.; Akita, H.; Kunii, T.; Shirai, R.; Takeda, T. Long-term oral administration of curcumin is effective in preventing short-term memory deterioration and prolonging lifespan in a mouse model of Alzheimer’s disease. Adv. Tradit. Med. 2023, 1–13. [Google Scholar] [CrossRef]
- Yan, D.D.; Wang, N.; Yao, J.L.; Wu, X.; Yuan, J.P.; Yan, H. Curcumin Attenuates the PERK-eIF2 alpha Signaling to Relieve Acrylamide-Induced Neurotoxicity in SH-SY5Y Neuroblastoma Cells. Neurochem. Res. 2022, 47, 1037–1048. [Google Scholar] [CrossRef]
- Bustanji, Y.; Taha, M.O.; Almasri, I.M.; Al-Ghussein, M.A.S.; Mohammad, M.K.; Alkhatib, H.S. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J. Enzym. Inhib. Med. Chem. 2009, 24, 771–778. [Google Scholar] [CrossRef]
- Hongo, N.; Takamura, Y.; Nishimaru, H.; Matsumoto, J.; Tobe, K.; Saito, T.; Saido, T.C.; Nishijo, H. Astaxanthin Ameliorated Parvalbumin-Positive Neuron Deficits and Alzheimer’s Disease-Related Pathological Progression in the Hippocampus of AppNL-G-F/NL-G-F Mice. Front. Pharmacol. 2020, 11, 15. [Google Scholar] [CrossRef]
- Che, H.X.; Li, Q.; Zhang, T.T.; Wang, D.D.; Yang, L.; Xu, J.; Yanagita, T.; Xue, C.H.; Chang, Y.G.; Wang, Y.M. Effects of Astaxanthin and Docosahexaenoic-Acid-Acylated Astaxanthin on Alzheimer’s Disease in APP/PS1 Double-Transgenic Mice. J. Agric. Food Chem. 2018, 66, 4948–4957. [Google Scholar] [CrossRef]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.B.; Rui, Y.H.; Qin, L.Q.; Xu, J.Y.; Han, S.F.; Yuan, L.X.; Yin, X.B.; Wan, Z.X. Vitamin D Combined with Resveratrol Prevents Cognitive Decline in SAMP8 Mice. Curr. Alzheimer Res. 2017, 14, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Resveratrol Ameliorates Tau Hyperphosphorylation at Ser396 Site and Oxidative Damage in Rat Hippocampal Slices Exposed to Vanadate: Implication of ERK1/2 and GSK-3/β Signaling Cascades. J. Agric. Food Chem. 2017, 65, 9626–9634. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A.; Alfaifi, M.Y. Trans-resveratrol Inhibits Tau Phosphorylation in the Brains of Control and Cadmium Chloride-Treated Rats by Activating PP2A and PI3K/Akt Induced-Inhibition of GSK3. Neurochem. Res. 2019, 44, 357–373. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Li, Z.H.; Rizak, J.D.; Wu, S.H.; Wang, Z.B.; He, R.Q.; Su, M.; Qin, D.D.; Wang, J.K.; Hu, X.T. Resveratrol Attenuates Formaldehyde Induced Hyperphosphorylation of Tau Protein and Cytotoxicity in N2a Cells. Front. Neurosci. 2017, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S.; Belai, A. Natural Therapies of the Inflammatory Bowel Disease: The Case of Rutin and its Aglycone, Quercetin. Mini-Rev. Med. Chem. 2018, 18, 234–243. [Google Scholar] [CrossRef]
- Negahdari, R.; Bohlouli, S.; Sharifi, S.; Dizaj, S.M.; Saadat, Y.R.; Khezri, K.; Jafari, S.; Ahmadian, E.; Jahandizi, N.G.; Raeesi, S. Therapeutic benefits of rutin and its nanoformulations. Phytother. Res. 2021, 35, 1719–1738. [Google Scholar] [CrossRef]
- Song, H.L.; Zhang, X.; Wang, W.Z.; Liu, R.H.; Zhao, K.; Liu, M.Y.; Gong, W.M.; Ning, B. Neuroprotective mechanisms of rutin for spinal cord injury through anti-oxidation and anti-inflammation and inhibition of p38 mitogen activated protein kinase pathway. Neural Regen. Res. 2018, 13, 128–134. [Google Scholar] [CrossRef]
- Sun, X.Y.; Li, L.J.; Dong, Q.X.; Zhu, J.; Huang, Y.R.; Hou, S.J.; Yu, X.L.; Liu, R.T. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 14. [Google Scholar] [CrossRef]
- Feligioni, M.; Brambilla, E.; Camassa, A.; Sclip, A.; Arnaboldi, A.; Morelli, F.; Antoniou, X.; Borsello, T. Crosstalk between JNK and SUMO Signaling Pathways: DeSUMOylation Is Protective against H2O2-Induced Cell Injury. PLoS ONE 2011, 6, e28185. [Google Scholar] [CrossRef]
- Buccarello, L.; Dragotto, J.; Iorio, F.; Hassanzadeh, K.; Corbo, M.; Feligioni, M. The pivotal role of SUMO-1-JNK-Tau axis in an in vitro model of oxidative stress counteracted by the protective effect of curcumin. Biochem. Pharmacol. 2020, 178, 114066. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; McCarty-Wood, E.; Van Deerlin, V.M.; Lee, V.M.Y.; Trojanowski, J.Q. Acetylated Tau Neuropathology in Sporadic and Hereditary Tauopathies. Am. J. Pathol. 2013, 183, 344–351. [Google Scholar] [CrossRef]
- Yan, J.; Luo, A.L.; Sun, R.; Tang, X.L.; Zhao, Y.L.; Zhang, J.; Zhou, B.Y.; Zheng, H.; Yu, H.H.; Li, S.Y. Resveratrol Mitigates Hippocampal Tau Acetylation and Cognitive Deficit by Activation SIRT1 in Aged Rats following Anesthesia and Surgery. Oxidative Med. Cell. Longev. 2020, 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimer’s Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Qi, B.T.; Tan, J.W.; Sun, Y.X.; Gong, Y.H.; Zhang, Q.W. Mechanistic insight into the disruption of Tau R3-R4 protofibrils by curcumin and epinephrine: An all-atom molecular dynamics study. Phys. Chem. Chem. Phys. 2022, 24, 20454–20465. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Zuo, X.H.; Yang, F.S.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.X.; Chen, P.P.; Maiti, P.; et al. Curcumin Suppresses Soluble Tau Dimers and Corrects Molecular Chaperone, Synaptic, and Behavioral Deficits in Aged Human Tau Transgenic Mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- El Mammeri, N.; Dregni, A.J.; Duan, P.; Wang, H.K.; Hong, M. Microtubule-binding core of the tau protein. Sci. Adv. 2022, 8, eabo4459. [Google Scholar] [CrossRef]
- Lo Cascio, F.; Puangmalai, N.; Ellsworth, A.; Bucchieri, F.; Pace, A.; Piccionello, A.P.; Kayed, R. Toxic Tau Oligomers Modulated by Novel Curcumin Derivatives. Sci. Rep. 2019, 9, 19011. [Google Scholar] [CrossRef]
- Bijari, N.; Balalaie, S.; Akbari, V.; Golmohammadi, F.; Moradi, S.; Adibi, H.; Khodarahmi, R. Effective suppression of the modified PHF6 peptide/1N4R Tau amyloid aggregation by intact curcumin, not its degradation products: Another evidence for the pigment as preventive/therapeutic “functional food”. Int. J. Biol. Macromol. 2018, 120, 1009–1022. [Google Scholar] [CrossRef]
- Gueroux, M.; Fleau, C.; Slozeck, M.; Laguerre, M.; Pianet, I. Epigallocatechin 3-Gallate as an Inhibitor of Tau Phosphorylation and Aggregation: A Molecular and Structural Insight. J. Prev. Alzheimer’s Dis. 2017, 4, 218–225. [Google Scholar] [CrossRef]
- Sonawane, S.K.; Chidambaram, H.; Boral, D.; Gorantla, N.V.; Balmik, A.A.; Dangi, A.; Ramasamy, S.; Marelli, U.K.; Chinnathambi, S. EGCG impedes human Tau aggregation and interacts with Tau. Sci. Rep. 2020, 10, 12579. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Siddiqui, M.A.; Khan, M.M.; Ajmal, M.; Ahsan, R.; Rahaman, M.A.; Ahmad, M.A.; Arshad, M.; Khushtar, M. Current Pharmacological Trends on Myricetin. Drug Res. 2020, 70, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Zhong, T.; Chen, Z.X.; Chen, W.; Zhang, N.; Liu, X.L.; Wang, L.Q.; Chen, J.; Liang, Y. Myricetin slows liquid-liquid phase separation of Tau and activates ATG5-dependent autophagy to suppress Tau toxicity. J. Biol. Chem. 2021, 297, 17. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, W.M.; Masunov, A.E. Atomistic mechanism of polyphenol amyloid aggregation inhibitors: Molecular dynamics study of Curcumin, Exifone, and Myricetin interaction with the segment of tau peptide oligomer. J. Biomol. Struct. Dyn. 2015, 33, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.H.; Han, Y.L.; Yan, X.; Guan, Y.X. Proanthocyanidins prevent tau protein aggregation and disintegrate tau filaments. Chin. J. Chem. Eng. 2023, 57, 63–71. [Google Scholar] [CrossRef]
- KrishnaKumar, V.G.; Paul, A.; Gazit, E.; Segal, D. Mechanistic insights into remodeled Tau-derived PHF6 peptide fibrils by Naphthoquinone-Tryptophan hybrids. Sci. Rep. 2018, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Liu, R.D.; Wang, R.K.; Xin, Y.J.; Wu, Z.T.; Ba, Y.; Zhang, H.Z.; Cheng, X.M.; Zhou, G.Y.; Huang, H. Attenuation of Pb-induced Aβ generation and autophagic dysfunction via activation of SIRT1: Neuroprotective properties of resveratrol. Ecotox. Environ. Safe. 2021, 222, 12. [Google Scholar] [CrossRef]
- Martin, M.D.; Calcul, L.; Smith, C.; Jinwal, U.K.; Fontaine, S.N.; Darling, A.; Seeley, K.; Wojtas, L.; Narayan, M.; Gestwicki, J.E.; et al. Synthesis, Stereochemical Analysis, and Derivatization of Myricanol Provide New Probes That Promote Autophagic Tau Clearance. ACS Chem. Biol. 2015, 10, 1099–1109. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Xiao, Q.L.; Yan, P.; Ma, X.C.; Liu, H.Y.; Perez, R.; Zhu, A.; Gonzales, E.; Tripoli, D.L.; Czerniewski, L.; Ballabio, A.; et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing A beta Generation and Amyloid Plaque Pathogenesis. J. Neurosci. 2015, 35, 12137–12151. [Google Scholar] [CrossRef]
- Polito, V.A.; Li, H.M.; Martini-Stoica, H.; Wang, B.P.; Yang, L.; Xu, Y.; Swartzlander, D.B.; Palmieri, M.; di Ronza, A.; Lee, V.M.Y.; et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014, 6, 1142–1160. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.B.; Tong, B.C.K.; Iyaswamy, A.; Shang, W.B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2020, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Corpas, R.; Griñán-Ferré, C.; Rodríguez-Farré, E.; Pallàs, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, J.G.; Li, H.; Yang, H.M. Pharmacological Effects of Active Components of Chinese Herbal Medicine in the Treatment of Alzheimer’s Disease: A Review. Am. J. Chin. Med. 2016, 44, 1525–1541. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Chen, J.J.; Bi, D.C.; Gu, L.; Yao, L.J.; Li, X.T.; Li, H.; Xu, H.; Hu, Z.L.; Liu, Q.; et al. Specific Degradation of Endogenous Tau Protein and Inhibition of Tau Fibrillation by Tanshinone IIA through the Ubiquitin-Proteasome Pathway. J. Agric. Food Chem. 2020, 68, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Broderick, T.L.; Rasool, S.; Li, R.Z.; Zhang, Y.X.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. [Google Scholar] [CrossRef]
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V.W. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31. [Google Scholar] [CrossRef]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef]
- Moll, A.; Ramirez, L.M.; Ninov, M.; Schwarz, J.; Urlaub, H.; Zweckstetter, M. Hsp multichaperone complex buffers pathologically modified Tau. Nat. Commun. 2022, 13, 3668. [Google Scholar] [CrossRef] [PubMed]
- Assaye, M.A.; Gizaw, S.T. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. Int. J. Gen. Med. 2022, 15, 5635–5649. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.L.; Kong, L.B.; Cao, Y.; Yan, L. Identification and Quantification, Metabolism and Pharmacokinetics, Pharmacological Activities, and Botanical Preparations of Protopine: A Review. Molecules 2022, 27, 215. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Senapati, S.; Malampati, S.; Zhu, Z.; Su, C.F.; Liu, J.; Guan, X.J.; Tong, B.C.K.; et al. Protopine promotes the proteasomal degradation of pathological tau in Alzheimer’s disease models via HDAC6 inhibition. Phytomedicine 2022, 96, 14. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Reddi, R.N.; Kammala, A.K.; Vasudevan, K.; Senapati, S.; Zhu, Z.; Su, C.F.; Liu, J.; et al. Bromo-protopine, a novel protopine derivative, alleviates tau pathology by activating chaperone-mediated autophagy for Alzheimer’s disease therapy. Front. Mol. Biosci. 2022, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Zarrabi, A.; Najafi, M.; Samarghandian, S.; Mohammadinejad, R.; Ahn, K.S. Resveratrol targeting tau proteins, amyloid-beta aggregations, and their adverse effects: An updated review. Phytother. Res. 2020, 34, 2867–2888. [Google Scholar] [CrossRef]
- Sato, R.; Vohra, S.; Yamamoto, S.; Suzuki, K.; Pavel, K.; Shulga, S.; Blume, Y.; Kurita, N. Specific interactions between tau protein and curcumin derivatives: Molecular docking and ab initio molecular orbital simulations. J. Mol. Graph. 2020, 98, 11. [Google Scholar] [CrossRef]
- Sivanantharajah, L.; Mudher, A. Curcumin as a Holistic Treatment for Tau Pathology. Front. Pharmacol. 2022, 13, 8. [Google Scholar] [CrossRef]
- Hamimed, S.; Jabberi, M.; Chatti, A. Nanotechnology in drug and gene delivery. Naunyn-Schmiedebergs Arch. Pharmacol. 2022, 395, 769–787. [Google Scholar] [CrossRef]
- Shabbir, U.; Rubab, M.; Tyagi, A.; Oh, D.H. Curcumin and Its Derivatives as Theranostic Agents in Alzheimer’s Disease: The Implication of Nanotechnology. Int. J. Mol. Sci. 2021, 22, 196. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, H.; Zhao, Y. Modulation of Tau Pathology in Alzheimer’s Disease by Dietary Bioactive Compounds. Int. J. Mol. Sci. 2024, 25, 831. https://doi.org/10.3390/ijms25020831
Shi H, Zhao Y. Modulation of Tau Pathology in Alzheimer’s Disease by Dietary Bioactive Compounds. International Journal of Molecular Sciences. 2024; 25(2):831. https://doi.org/10.3390/ijms25020831
Chicago/Turabian StyleShi, Huahua, and Yan Zhao. 2024. "Modulation of Tau Pathology in Alzheimer’s Disease by Dietary Bioactive Compounds" International Journal of Molecular Sciences 25, no. 2: 831. https://doi.org/10.3390/ijms25020831
APA StyleShi, H., & Zhao, Y. (2024). Modulation of Tau Pathology in Alzheimer’s Disease by Dietary Bioactive Compounds. International Journal of Molecular Sciences, 25(2), 831. https://doi.org/10.3390/ijms25020831