

Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases
 ,
, 
Abstract
:1. Introduction
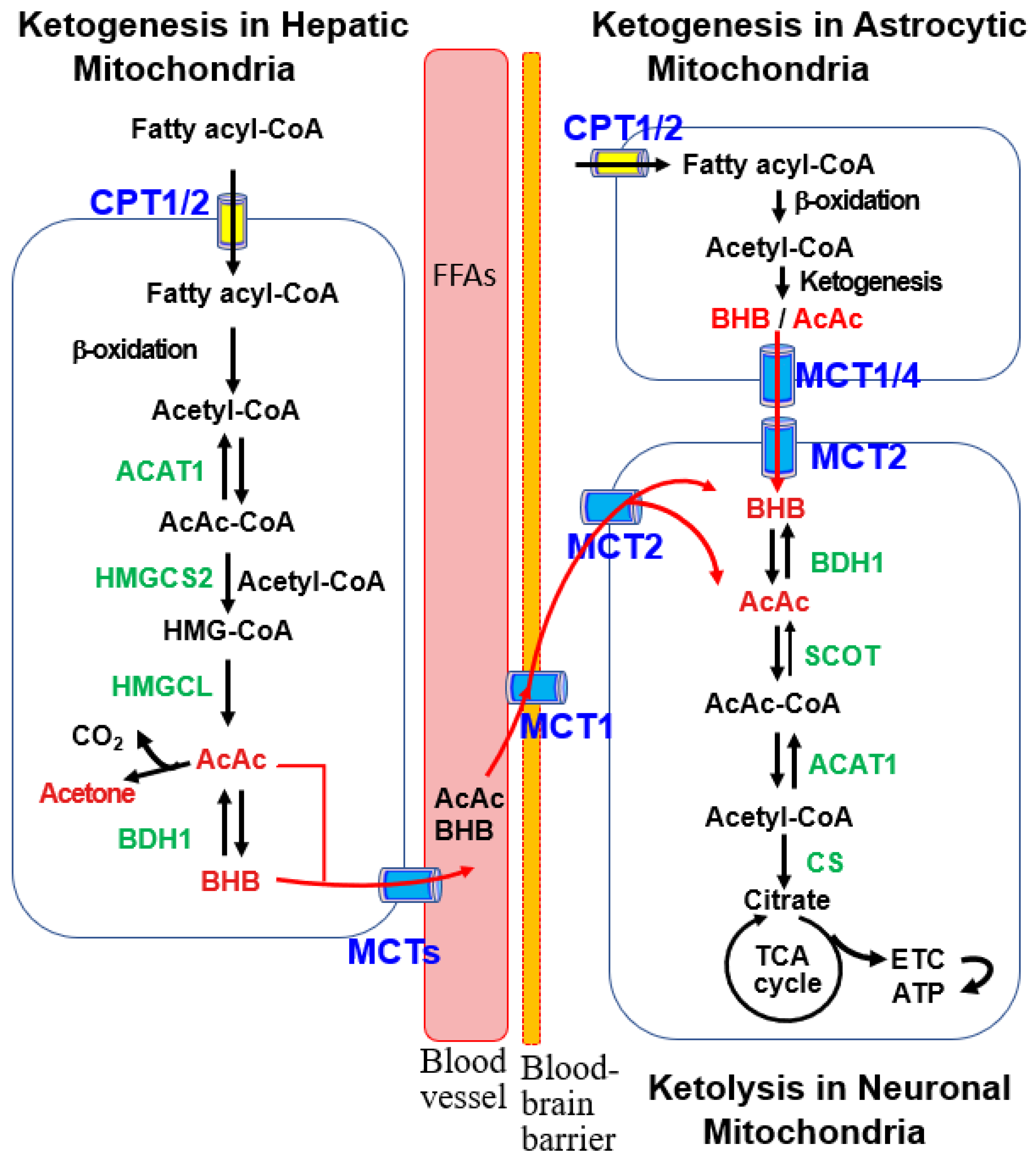
2. Ketone Body Metabolism in the Brain
2.1. Brain KBs Supplied by Ketogenesis in Liver and Astrocytes
2.2. Ketolysis in the Brain
3. Therapeutic Potential of KBs and KD in Cerebral Ischemia and Neurodegenerative Diseases
3.1. Cerebral Ischemia
3.2. Alzheimer’s Disease
3.3. Parkinson’s Disease
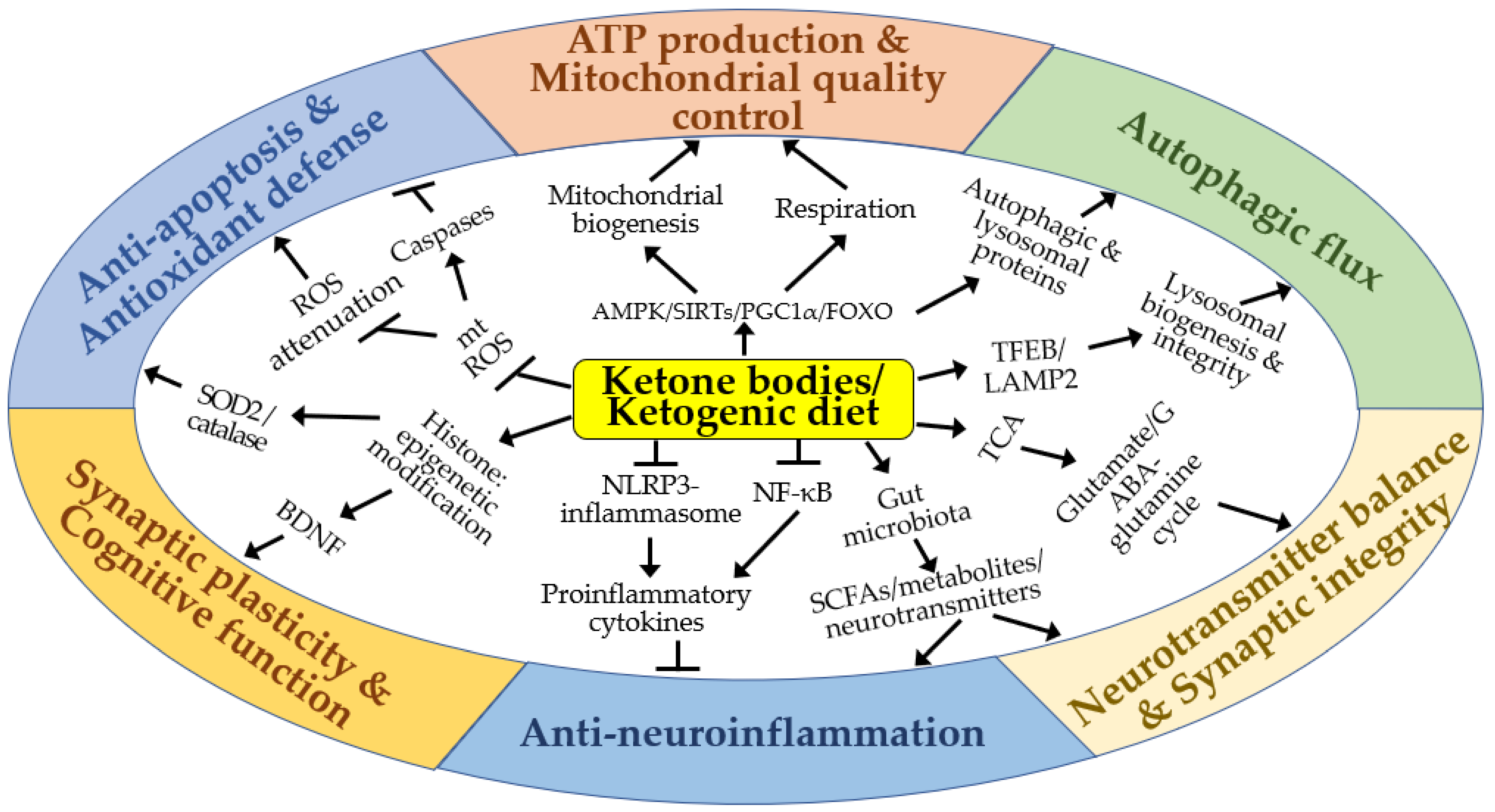
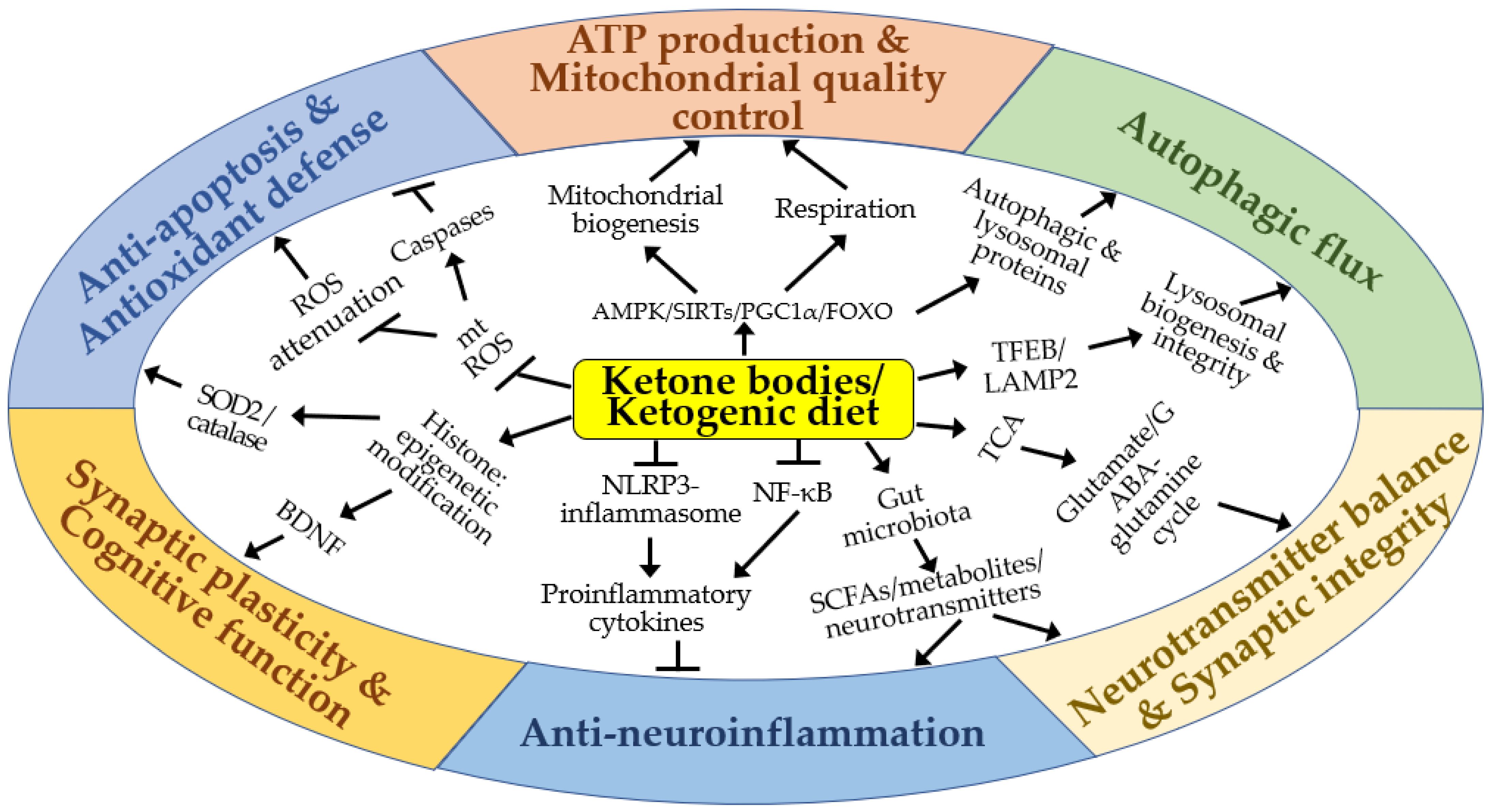
4. Molecular Mechanisms of the Neuroprotective Effects of KBs and KD
4.1. KBs as an Energy Source for the Brain
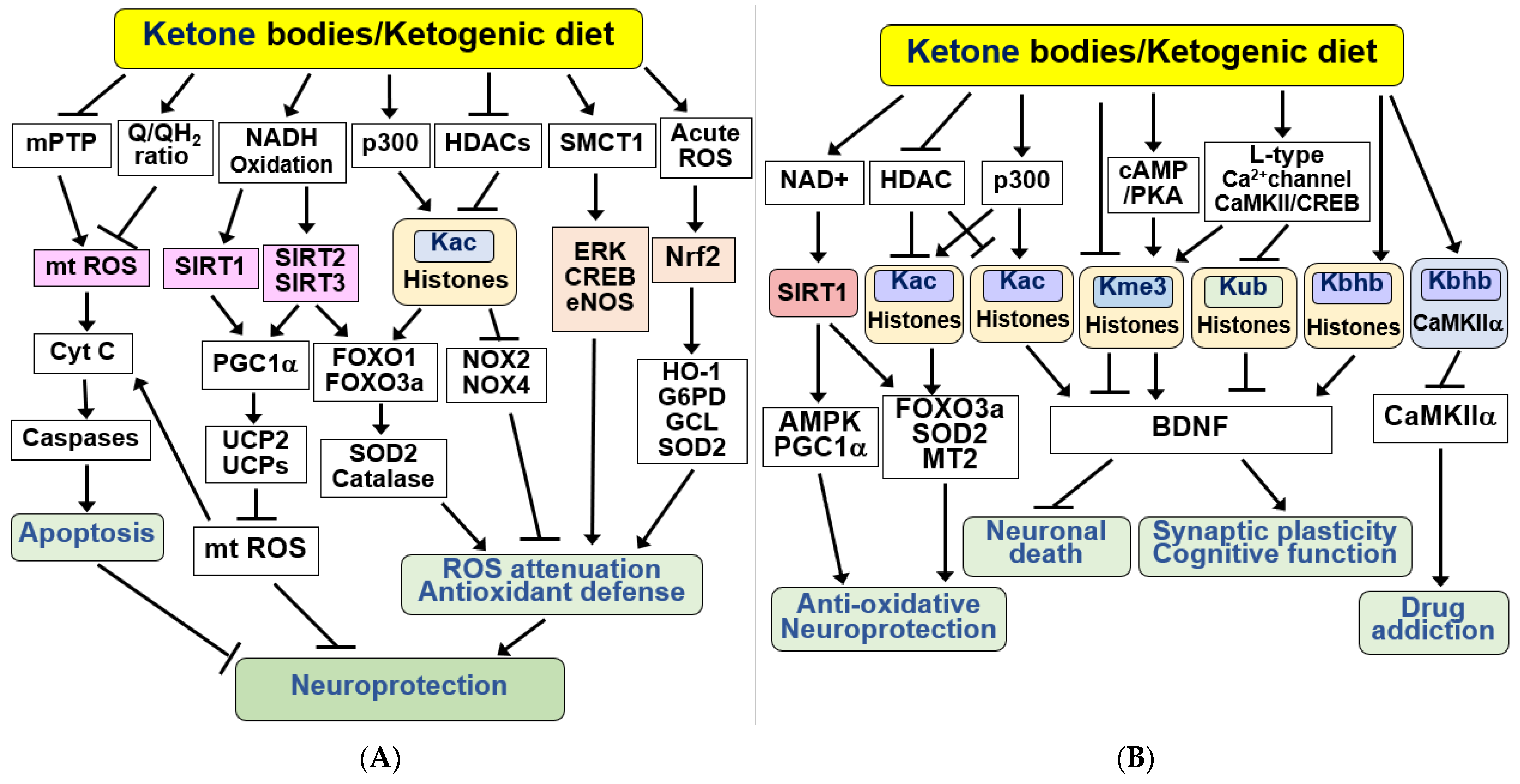
4.2. Beneficial Effects of KBs and KD on Mitochondrial Function
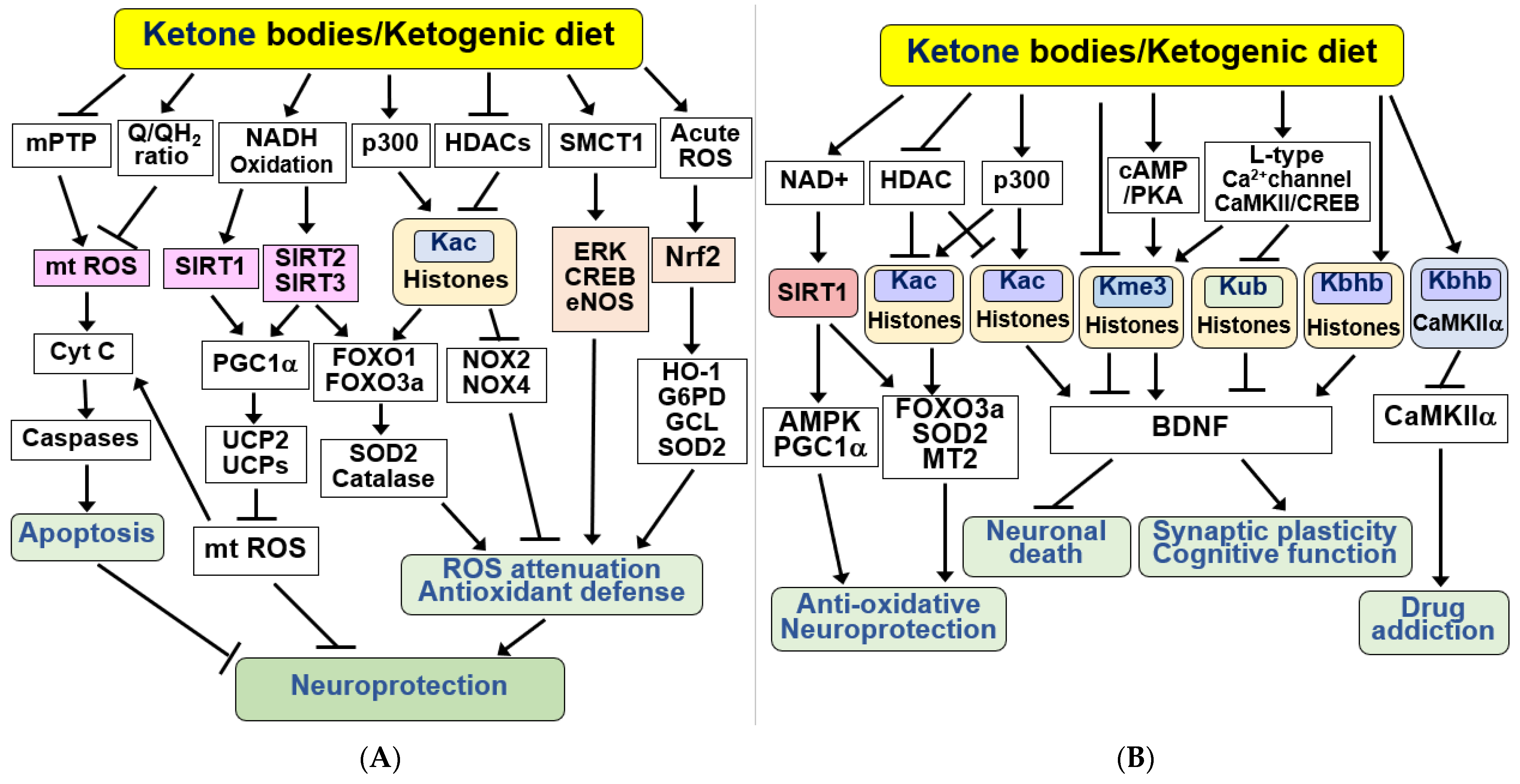
4.3. Anti-Apoptotic and Antioxidant Effects of KBs and KD in the Brain
4.4. KBs and KD as Epigenetic and Post-Translational Modifiers of Histones and Non-Histone Proteins
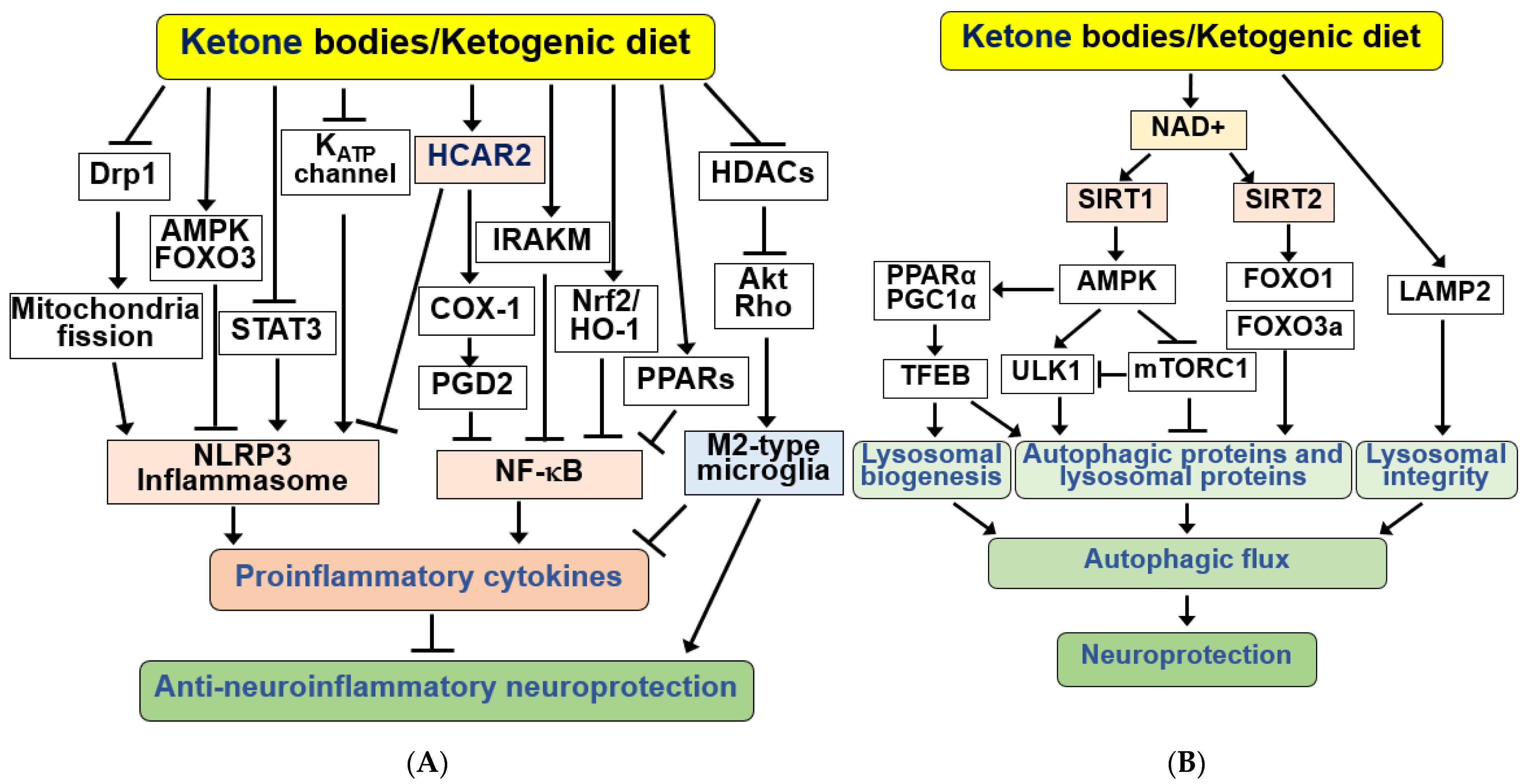
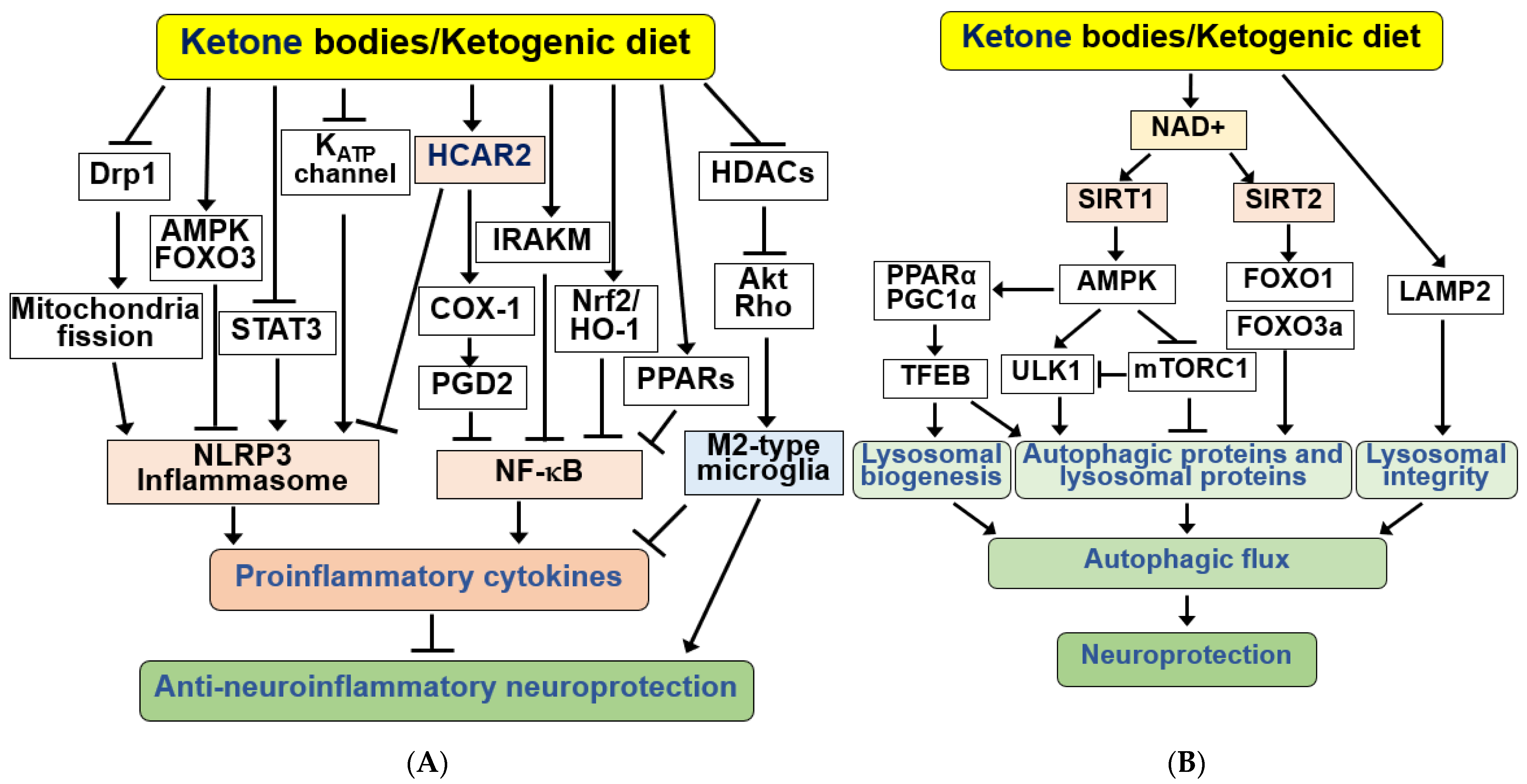
4.5. KBs and KD as Anti-Neuroinflammatory Signaling Modulators in the Brain
4.6. KBs and KD as Regulators of Autophagy
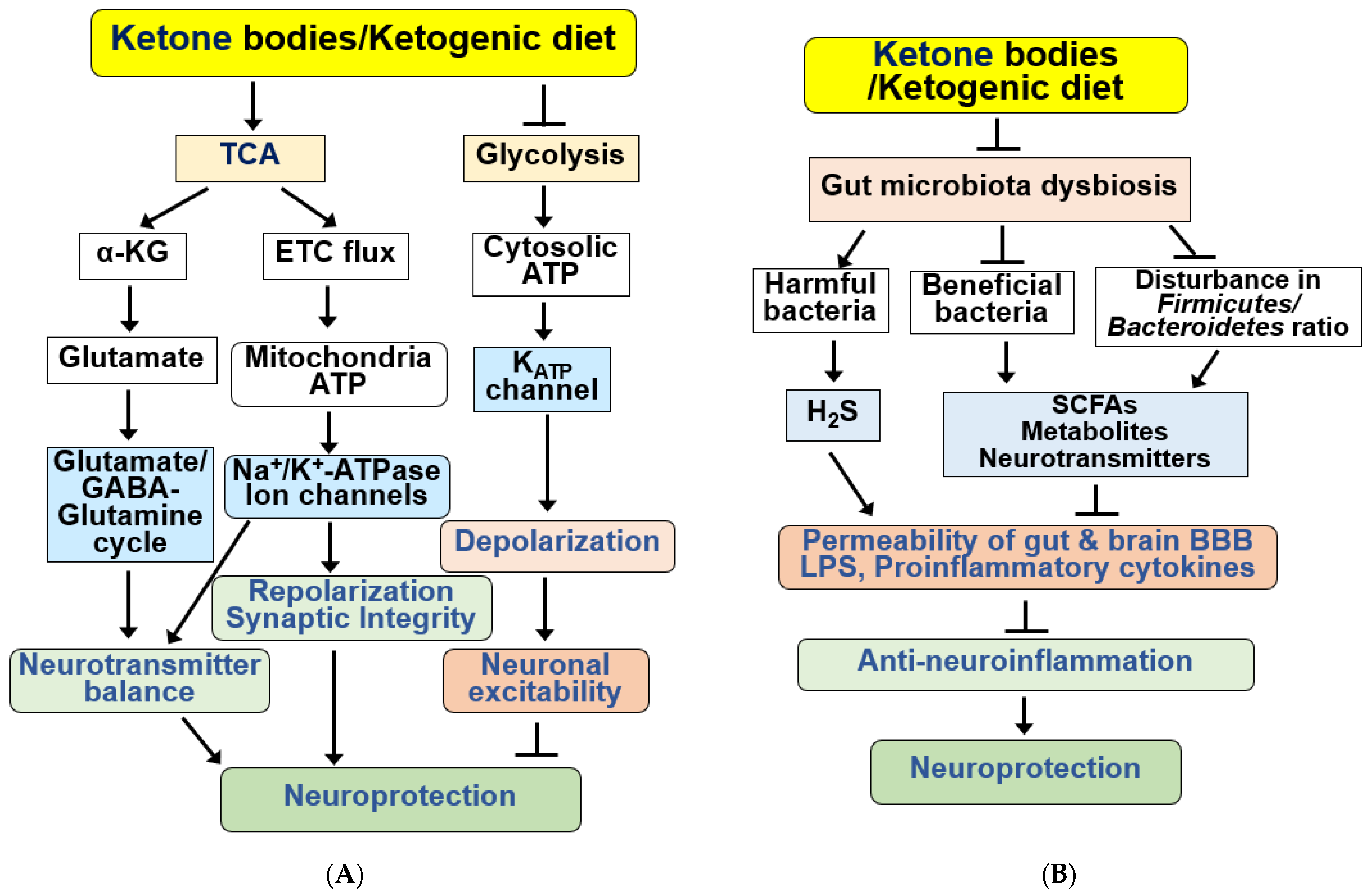
4.7. KBs and KD as Modulators of Neurotransmission Systems
4.8. KBs and KD as Regulators of the Gut Microbiota
4.9. Adverse Effects of the KD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Metabolic and Signaling Roles of Ketone Bodies in Health and Disease. Annu. Rev. Nutr. 2021, 41, 49–77. [Google Scholar] [CrossRef]
- Pietrzak, D.; Kasperek, K.; Rekawek, P.; Piatkowska-Chmiel, I. The Therapeutic Role of Ketogenic Diet in Neurological Disorders. Nutrients 2022, 14, 1952. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: A comprehensive review of the literature. Obes. Rev. 2020, 21, e13024. [Google Scholar] [CrossRef] [PubMed]
- Wlodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Abdul Kadir, A.; Clarke, K.; Evans, R.D. Cardiac ketone body metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165739. [Google Scholar] [CrossRef]
- Dabek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef]
- Tozzi, R.; Cipriani, F.; Masi, D.; Basciani, S.; Watanabe, M.; Lubrano, C.; Gnessi, L.; Mariani, S. Ketone Bodies and SIRT1, Synergic Epigenetic Regulators for Metabolic Health: A Narrative Review. Nutrients 2022, 14, 3145. [Google Scholar] [CrossRef]
- Wang, L.; Chen, P.; Xiao, W. beta-hydroxybutyrate as an Anti-Aging Metabolite. Nutrients 2021, 13, 3420. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Choe, W.; Yoon, K.S.; Ha, J.; Kim, S.S.; Yeo, E.J.; Kang, I. Molecular Mechanisms for Ketone Body Metabolism, Signaling Functions, and Therapeutic Potential in Cancer. Nutrients 2022, 14, 4932. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L. Cerebral ketone metabolism during development and injury. Epilepsy Res. 2012, 100, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Blazquez, C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014, 6, 1759091414550997. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Xu, D.; Zhou, Y.; Qu, Z.; Yang, Q.; Lv, Q. Low carbohydrate ketogenic diets reduce cardiovascular risk factor levels in obese or overweight patients with T2DM: A meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1092031. [Google Scholar] [CrossRef] [PubMed]
- Risi, R.; Tozzi, R.; Watanabe, M. Beyond weight loss in nonalcoholic fatty liver disease: The role of carbohydrate restriction. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients With Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bi, D.; Zhang, Y.; Kong, C.; Du, J.; Wu, X.; Wei, Q.; Qin, H. Ketogenic diet for human diseases: The underlying mechanisms and potential for clinical implementations. Signal Transduct. Target. Ther. 2022, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Dynka, D.; Kowalcze, K.; Paziewska, A. The Role of Ketogenic Diet in the Treatment of Neurological Diseases. Nutrients 2022, 14, 5003. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-beta-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s Disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.S.; Lin, S.J.; Liao, P.Y.; Suresh, D.; Hsu, T.R.; Wang, P.Y. Role of Ketogenic Diets in Multiple Sclerosis and Related Animal Models: An Updated Review. Adv. Nutr. 2022, 13, 2002–2014. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef]
- Makievskaya, C.I.; Popkov, V.A.; Andrianova, N.V.; Liao, X.; Zorov, D.B.; Plotnikov, E.Y. Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 2576. [Google Scholar] [CrossRef]
- Brenton, J.N.; Banwell, B.; Bergqvist, A.G.C.; Lehner-Gulotta, D.; Gampper, L.; Leytham, E.; Coleman, R.; Goldman, M.D. Pilot study of a ketogenic diet in relapsing-remitting MS. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e565. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; McManus, E.J.; Brinkhuis, M.; Romero-Ferrando, B. Time-Restricted Ketogenic Diet in Huntington’s Disease: A Case Study. Front. Behav. Neurosci. 2022, 16, 931636. [Google Scholar] [CrossRef] [PubMed]
- de la Rubia Orti, J.E.; Fernandez, D.; Platero, F.; Garcia-Pardo, M.P. Can Ketogenic Diet Improve Alzheimer’s Disease? Association with Anxiety, Depression, and Glutamate System. Front. Nutr. 2021, 8, 744398. [Google Scholar] [CrossRef] [PubMed]
- Dietch, D.M.; Kerr-Gaffney, J.; Hockey, M.; Marx, W.; Ruusunen, A.; Young, A.H.; Berk, M.; Mondelli, V. Efficacy of low carbohydrate and ketogenic diets in treating mood and anxiety disorders: Systematic review and implications for clinical practice. BJPsych Open 2023, 9, e70. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.W.Y.; Corley, M.J.; Pang, A.; Arakaki, G.; Abbott, L.; Nishimoto, M.; Miyamoto, R.; Lee, E.; Yamamoto, S.; Maunakea, A.K.; et al. A modified ketogenic gluten-free diet with MCT improves behavior in children with autism spectrum disorder. Physiol. Behav. 2018, 188, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rae, C.D. beta-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Moller, N. Ketone Body, 3-Hydroxybutyrate: Minor Metabolite—Major Medical Manifestations. J. Clin. Endocrinol. Metab. 2020, 105, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Kaviyarasan, S.; Chung Sia, E.L.; Retinasamy, T.; Arulsamy, A.; Shaikh, M.F. Regulation of gut microbiome by ketogenic diet in neurodegenerative diseases: A molecular crosstalk. Front. Aging Neurosci. 2022, 14, 1015837. [Google Scholar] [CrossRef]
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M.; Berk, M.; Carvalho, A.F.; Puri, B.K. Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 2020, 63, e8. [Google Scholar] [CrossRef]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Placais, P.Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef]
- Koch, K.; Berressem, D.; Konietzka, J.; Thinnes, A.; Eckert, G.P.; Klein, J. Hepatic Ketogenesis Induced by Middle Cerebral Artery Occlusion in Mice. J. Am. Heart Assoc. 2017, 6, e005556. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Suzuki, M.; Kitamura, Y.; Mori, S.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn. J. Pharmacol. 2002, 89, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Woods, A.; de Ceballos, M.L.; Carling, D.; Guzman, M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Longo, R.; Peri, C.; Cricri, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sun, L.; Wang, H. Ketogenic Diet for Neonatal Hypoxic-Ischemic Encephalopathy. ACS Chem. Neurosci. 2023, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Giza, C.C. Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev. Neurosci. 2006, 28, 447–456. [Google Scholar] [CrossRef]
- Koppel, S.J.; Pei, D.; Wilkins, H.M.; Weidling, I.W.; Wang, X.; Menta, B.W.; Perez-Ortiz, J.; Kalani, A.; Manley, S.; Novikova, L.; et al. A ketogenic diet differentially affects neuron and astrocyte transcription. J. Neurochem. 2021, 157, 1930–1945. [Google Scholar] [CrossRef]
- Saito, E.R.; Miller, J.B.; Harari, O.; Cruchaga, C.; Mihindukulasuriya, K.A.; Kauwe, J.S.K.; Bikman, B.T. Alzheimer’s disease alters oligodendrocytic glycolytic and ketolytic gene expression. Alzheimers Dement. 2021, 17, 1474–1486. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Mintz, A.; Whitlow, C.; Jung, Y.; Solingapuram Sai, K.; Register, T.C.; Kellar, D.; Lockhart, S.N.; Hoscheidt, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.; Castellano, C.A.; St-Pierre, V.; Myette-Cote, E.; Langlois, F.; Roy, M.; Morin, M.C.; Bocti, C.; Fulop, T.; Godin, J.P.; et al. A ketogenic drink improves cognition in mild cognitive impairment: Results of a 6-month RCT. Alzheimers Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Y.; Zhang, X.; Liu, L.; Zhou, B.; Mo, R.; Li, Y.; Li, H.; Li, F.; Tao, Y.; et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4(-/-): A double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr. 2020, 39, 2092–2105. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef]
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Krikorian, R.; Shidler, M.D.; Summer, S.S.; Sullivan, P.G.; Duker, A.P.; Isaacson, R.S.; Espay, A.J. Nutritional ketosis for mild cognitive impairment in Parkinson’s disease: A controlled pilot trial. Clin. Park. Relat. Disord. 2019, 1, 41–47. [Google Scholar] [CrossRef]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain Ischemia as a Prelude to Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 636653. [Google Scholar] [CrossRef] [PubMed]
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Milaganur Mohan, H.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral ischemia induces the aggregation of proteins linked to neurodegenerative diseases. Sci. Rep. 2018, 8, 2701. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Zechel, J.L.; Valerio, J.; Emancipator, D.S.; Xu, K.; Pundik, S.; LaManna, J.C.; Lust, W.D. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Ye, L.; Sharma, K.; Jin, Y.; Harrison, M.M.; Caldwell, T.; Berthiaume, J.M.; Luo, Y.; LaManna, J.C.; Puchowicz, M.A. Diet-Induced Ketosis Protects Against Focal Cerebral Ischemia in Mouse. Adv. Exp. Med. Biol. 2017, 977, 205–213. [Google Scholar] [CrossRef]
- Lehto, A.; Koch, K.; Barnstorf-Brandes, J.; Viel, C.; Fuchs, M.; Klein, J. ss-Hydroxybutyrate Improves Mitochondrial Function After Transient Ischemia in the Mouse. Neurochem. Res. 2022, 47, 3241–3249. [Google Scholar] [CrossRef]
- Shaafi, S.; Sharifi-Bonab, M.; Ghaemian, N.; Mokhtarkhani, M.; Akbari, H. Early Motor-Behavioral Outcome of Ischemic Stroke with Ketogenic Diet Preconditioning: Interventional Animal Study. J. Stroke Cerebrovasc. Dis. 2019, 28, 1032–1039. [Google Scholar] [CrossRef]
- Lin, C.; Wang, S.; Xie, J.; Zhu, J.; Xu, J.; Liu, K.; Chen, J.; Yu, M.; Zhong, H.; Huang, K.; et al. Ketogenic diet and beta-Hydroxybutyrate alleviate ischemic brain injury in mice via an IRAKM-dependent pathway. Eur. J. Pharmacol. 2023, 955, 175933. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.K.; Nguyen, N.; Pham, L.; Truong, D.D. Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J. Neural Transm. 2008, 115, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e21788. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A Dietary Ketone Ester Normalizes Abnormal Behavior in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1044. [Google Scholar] [CrossRef]
- Beckett, T.L.; Studzinski, C.M.; Keller, J.N.; Paul Murphy, M.; Niedowicz, D.M. A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1505, 61–67. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Puri, B.K.; Carvalho, A.; Maes, M.; Berk, M.; Ruusunen, A.; Olive, L. Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought? Int. J. Neuropsychopharmacol. 2020, 23, 366–384. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yin, X.; Wang, M.; Wang, Y.; Li, F.; Gao, Y.; Han, G.; Gao, Z.; Wang, Z. beta-Hydroxybutyrate alleviates pyroptosis in MPP(+)/MPTP-induced Parkinson’s disease models via inhibiting STAT3/NLRP3/GSDMD pathway. Int. Immunopharmacol. 2022, 113, 109451. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef]
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X.; Liu, S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 2009, 1286, 25–31. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Avgerinos, K.I. Brain glucose and ketone utilization in brain aging and neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 154, 79–110. [Google Scholar] [CrossRef]
- Myette-Cote, E.; Soto-Mota, A.; Cunnane, S.C. Ketones: Potential to achieve brain energy rescue and sustain cognitive health during ageing. Br. J. Nutr. 2022, 128, 407–423. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Svart, M.; Gormsen, L.C.; Hansen, J.; Zeidler, D.; Gejl, M.; Vang, K.; Aanerud, J.; Moeller, N. Regional cerebral effects of ketone body infusion with 3-hydroxybutyrate in humans: Reduced glucose uptake, unchanged oxygen consumption and increased blood flow by positron emission tomography. A randomized, controlled trial. PLoS ONE 2018, 13, e0190556. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Poff, A.M.; Moss, S.; Soliven, M.; D’Agostino, D.P. Ketone Supplementation: Meeting the Needs of the Brain in an Energy Crisis. Front. Nutr. 2021, 8, 783659. [Google Scholar] [CrossRef] [PubMed]
- Mujica-Parodi, L.R.; Amgalan, A.; Sultan, S.F.; Antal, B.; Sun, X.; Skiena, S.; Lithen, A.; Adra, N.; Ratai, E.M.; Weistuch, C.; et al. Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc. Natl. Acad. Sci. USA 2020, 117, 6170–6177. [Google Scholar] [CrossRef]
- Sultana, M.A.; Hia, R.A.; Akinsiku, O.; Hegde, V. Peripheral Mitochondrial Dysfunction: A Potential Contributor to the Development of Metabolic Disorders and Alzheimer’s Disease. Biology 2023, 12, 1019. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Jaramillo, J.G.; Clarke, K.; Soto, A. Ketotherapeutics for neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 155, 141–168. [Google Scholar] [CrossRef]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef]
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.W.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J.H. The ketogenic diet as a therapeutic intervention strategy in mitochondrial disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050. [Google Scholar] [CrossRef]
- Gureev, A.P.; Silachev, D.N.; Sadovnikova, I.S.; Krutskikh, E.P.; Chernyshova, E.V.; Volodina, D.E.; Samoylova, N.A.; Potanina, D.V.; Burakova, I.Y.; Smirnova, Y.D.; et al. The Ketogenic Diet but not Hydroxycitric Acid Keeps Brain Mitochondria Quality Control and mtDNA Integrity Under Focal Stroke. Mol. Neurobiol. 2023, 60, 4288–4303. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.; Ruskin, D.N.; Sacchetti, P.; Masino, S.A. A unifying mechanism of ketogenic diet action: The multiple roles of nicotinamide adenine dinucleotide. Epilepsy Res. 2020, 167, 106469. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Tishkoff, D.X.; Bao, J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb. Exp. Pharmacol. 2011, 206, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1alpha-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Wang, S.J.; Zhao, X.H.; Chen, W.; Bo, N.; Wang, X.J.; Chi, Z.F.; Wu, W. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 2015, 11, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Gomora-Garcia, J.C.; Montiel, T.; Huttenrauch, M.; Salcido-Gomez, A.; Garcia-Velazquez, L.; Ramiro-Cortes, Y.; Gomora, J.C.; Castro-Obregon, S.; Massieu, L. Effect of the Ketone Body, D-beta-Hydroxybutyrate, on Sirtuin2-Mediated Regulation of Mitochondrial Quality Control and the Autophagy-Lysosomal Pathway. Cells 2023, 12, 486. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 2015, 85, 631–639. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Dabke, P.; Das, A.M. Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta-Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons. Nutrients 2020, 12, 2379. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Leng, S.X.; Zhang, H. Ketogenic Diet: An Effective Treatment Approach for Neurodegenerative Diseases. Curr. Neuropharmacol. 2022, 20, 2303–2319. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Balietti, M.; Giorgetti, B.; Di Stefano, G.; Casoli, T.; Platano, D.; Solazzi, M.; Bertoni-Freddari, C.; Aicardi, G.; Lattanzio, F.; Fattoretti, P. A ketogenic diet increases succinic dehydrogenase (SDH) activity and recovers age-related decrease in numeric density of SDH-positive mitochondria in cerebellar Purkinje cells of late-adult rats. Micron 2010, 41, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Haces, M.L.; Hernandez-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Davis, L.M.; Sullivan, P.G.; Maalouf, M.; Simeone, T.A.; van Brederode, J.; Rho, J.M. Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 2007, 101, 1316–1326. [Google Scholar] [CrossRef]
- Kong, G.; Huang, Z.; Ji, W.; Wang, X.; Liu, J.; Wu, X.; Huang, Z.; Li, R.; Zhu, Q. The Ketone Metabolite beta-Hydroxybutyrate Attenuates Oxidative Stress in Spinal Cord Injury by Suppression of Class I Histone Deacetylases. J. Neurotrauma 2017, 34, 2645–2655. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chan, S.L.; de Souza-Pinto, N.C.; Slevin, J.R.; Wersto, R.P.; Zhan, M.; Mustafa, K.; de Cabo, R.; Mattson, M.P. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 2006, 8, 389–414. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Karthikeyan, A.; Lu, J.; Ling, E.A.; Dheen, S.T. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 2015, 311, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox. Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, X.; Zhang, Q.; Li, Z.; Ma, S.; Bao, J.; Li, Z.; Bai, X.; Zheng, L.; Zhang, Z.; et al. PGC-1alpha/ERRalpha-Sirt3 Pathway Regulates DAergic Neuronal Death by Directly Deacetylating SOD2 and ATP Synthase beta. Antioxid. Redox Signal. 2016, 24, 312–328. [Google Scholar] [CrossRef]
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “great” controlling nucleotide coenzymes. IUBMB Life 2019, 71, 565–579. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Ma, A.; Kang, Y. Rational Application of beta-Hydroxybutyrate Attenuates Ischemic Stroke by Suppressing Oxidative Stress and Mitochondrial-Dependent Apoptosis via Activation of the Erk/CREB/eNOS Pathway. ACS Chem. Neurosci. 2021, 12, 1219–1227. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Rohling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed]
- Izuta, Y.; Imada, T.; Hisamura, R.; Oonishi, E.; Nakamura, S.; Inagaki, E.; Ito, M.; Soga, T.; Tsubota, K. Ketone body 3-hydroxybutyrate mimics calorie restriction via the Nrf2 activator, fumarate, in the retina. Aging Cell 2018, 17, e12699. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Bai, D.; Tang, M.; Zhang, M.; Zhao, L.; Li, J.; Yang, R.; Jiang, G. Ketogenic diet alleviates brain iron deposition and cognitive dysfunction via Nrf2-mediated ferroptosis pathway in APP/PS1 mouse. Brain Res. 2023, 1812, 148404. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yang, Y.; Zhang, B.; Han, G.; Yu, J.; Yu, Q.; Zhang, L. Ketone Body beta-Hydroxybutyric Acid Ameliorates Dopaminergic Neuron Injury Through Modulating Zinc Finger Protein 36/Acyl-CoA Synthetase Long-Chain Family Member Four Signaling Axis-Mediated Ferroptosis. Neuroscience 2023, 509, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Nielsen, M.; Li, S.; Shi, J. Ketones improves Apolipoprotein E4-related memory deficiency via sirtuin 3. Aging 2019, 11, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, A.; Li, Y.J.; Yang, Y.; Kishimoto, Y.; Zhang, S.; Wang, Y.; Wan, R.; Raefsky, S.M.; Lu, D.; et al. SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Commun. 2019, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer’s Disease Model. J. Neurosci. 2020, 40, 694–709. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone Bodies in Neurological Diseases: Focus on Neuroprotection and Underlying Mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef]
- Moreno, C.L.; Mobbs, C.V. Epigenetic mechanisms underlying lifespan and age-related effects of dietary restriction and the ketogenic diet. Mol. Cell. Endocrinol. 2017, 455, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, P.; Nettore, I.C.; Franchini, F.; Palatucci, G.; Muscogiuri, G.; Colao, A.; Macchia, P.E. Epigenome Modulation Induced by Ketogenic Diets. Nutrients 2022, 14, 3245. [Google Scholar] [CrossRef] [PubMed]
- Bandera-Merchan, B.; Boughanem, H.; Crujeiras, A.B.; Macias-Gonzalez, M.; Tinahones, F.J. Ketotherapy as an epigenetic modifier in cancer. Rev. Endocr. Metab. Disord. 2020, 21, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhan, Z.; Zhang, J.; Zhou, F.; An, L. Beta-Hydroxybutyrate Ameliorates Abeta-Induced Downregulation of TrkA Expression by Inhibiting HDAC1/3 in SH-SY5Y Cells. Am. J. Alzheimers Dis. Other Demen. 2020, 35, 1533317519883496. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Huang, X.; Cheng, X.; Lin, X.; Zhao, T.; Wu, L.; Yu, X.; Wu, K.; Fan, M.; Zhu, L. Ketogenic diet improves the spatial memory impairment caused by exposure to hypobaric hypoxia through increased acetylation of histones in rats. PLoS ONE 2017, 12, e0174477. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Simeone, K.A.; Stafstrom, C.E.; Rho, J.M. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 2018, 133, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Yang, D.; Ni, H.Y.; Xu, X.M.; Wu, F.; Lin, L.; Chen, J.; Sun, Y.Y.; Huang, Z.Q.; Li, S.Y.; et al. Ketone bodies promote stroke recovery via GAT-1-dependent cortical network remodeling. Cell Rep. 2023, 42, 112294. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Murugan, M.; Boison, D. Ketogenic diet, neuroprotection, and antiepileptogenesis. Epilepsy Res. 2020, 167, 106444. [Google Scholar] [CrossRef]
- Chen, F.; He, X.; Luan, G.; Li, T. Role of DNA Methylation and Adenosine in Ketogenic Diet for Pharmacoresistant Epilepsy: Focus on Epileptogenesis and Associated Comorbidities. Front. Neurol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Du, H.; Shang, S.; Zhang, Y.; Lu, X. Beta-Hydroxybutyrate Enhances BDNF Expression by Increasing H3K4me3 and Decreasing H2AK119ub in Hippocampal Neurons. Front. Neurosci. 2020, 14, 591177. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Du, H.; Zhu, X.; Wang, L.; Shang, S.; Wu, X.; Lu, H.; Lu, X. Beta-hydroxybutyrate Promotes the Expression of BDNF in Hippocampal Neurons under Adequate Glucose Supply. Neuroscience 2018, 386, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Cheng, X.; He, Y.; Xie, Y.; Xu, F.; Xu, Y.; Huang, W. Function and mechanism of histone beta-hydroxybutyrylation in health and disease. Front. Immunol. 2022, 13, 981285. [Google Scholar] [CrossRef] [PubMed]
- Cornuti, S.; Chen, S.; Lupori, L.; Finamore, F.; Carli, F.; Samad, M.; Fenizia, S.; Caldarelli, M.; Damiani, F.; Raimondi, F.; et al. Brain histone beta-hydroxybutyrylation couples metabolism with gene expression. Cell Mol. Life Sci. 2023, 80, 28. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Chen, L.; Miao, Z.; Xu, X. beta-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-beta-hydroxybutyrylation. Biochem. Biophys. Res. Commun. 2017, 490, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wan, X.; Wu, Z.; Zhou, Y.; Chen, R.; Xu, W.; Zhang, J.; Yang, Z.; Bai, L.; Zhang, J.; et al. beta-hydroxybutyrate reduces reinstatement of cocaine conditioned place preference through hippocampal CaMKII-alpha beta-hydroxybutyrylation. Cell Rep. 2022, 41, 111724. [Google Scholar] [CrossRef]
- Jayashankar, S.S.; Tajul Arifin, K.; Nasaruddin, M.L. beta-Hydroxybutyrate Regulates Activated Microglia to Alleviate Neurodegenerative Processes in Neurological Diseases: A Scoping Review. Nutrients 2023, 15, 524. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Taing, K.; Chen, L.; Weng, H.R. Emerging roles of GPR109A in regulation of neuroinflammation in neurological diseases and pain. Neural Regen. Res. 2023, 18, 763–768. [Google Scholar] [CrossRef]
- Jiang, Z.; Yin, X.; Wang, M.; Chen, T.; Wang, Y.; Gao, Z.; Wang, Z. Effects of Ketogenic Diet on Neuroinflammation in Neurodegenerative Diseases. Aging Dis. 2022, 13, 1146–1165. [Google Scholar] [CrossRef]
- Koh, S.; Dupuis, N.; Auvin, S. Ketogenic diet and Neuroinflammation. Epilepsy Res. 2020, 167, 106454. [Google Scholar] [CrossRef]
- Offermanns, S. Free fatty acid (FFA) and hydroxy carboxylic acid (HCA) receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 407–434. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef]
- Lukasova, M.; Malaval, C.; Gille, A.; Kero, J.; Offermanns, S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J. Clin. Investig. 2011, 121, 1163–1173. [Google Scholar] [CrossRef]
- Fu, S.P.; Li, S.N.; Wang, J.F.; Li, Y.; Xie, S.S.; Xue, W.J.; Liu, H.M.; Huang, B.X.; Lv, Q.K.; Lei, L.C.; et al. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-kappaB activation. Mediat. Inflamm. 2014, 2014, 983401. [Google Scholar] [CrossRef]
- Jiang, J.; Pan, H.; Shen, F.; Tan, Y.; Chen, S. Ketogenic diet alleviates cognitive dysfunction and neuroinflammation in APP/PS1 mice via the Nrf2/HO-1 and NF-kappaB signaling pathways. Neural Regen. Res. 2023, 18, 2767–2772. [Google Scholar] [CrossRef]
- Cullingford, T.E. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot Essent Fat. Acids 2004, 70, 253–264. [Google Scholar] [CrossRef]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-gamma activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gong, Y.; Luan, Y.; Li, Y.; Liu, J.; Yue, Z.; Yuan, B.; Sun, J.; Xie, C.; Li, L.; et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 1412–1429. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jiang, C.; Wu, J.; Liu, P.; Deng, X.; Zhang, Y.; Peng, B.; Zhu, Y. Ketogenic diet ameliorates cognitive impairment and neuroinflammation in a mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2022, 28, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.C.; Maisto, R.; Guida, F.; Boccella, S.; Luongo, L.; Balta, C.; D’Amico, G.; Herman, H.; Hermenean, A.; Bucolo, C.; et al. The activation of retinal HCA2 receptors by systemic beta-hydroxybutyrate inhibits diabetic retinal damage through reduction of endoplasmic reticulum stress and the NLRP3 inflammasome. PLoS ONE 2019, 14, e0211005. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. beta-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, X.; Zhao, Y.; Yang, Q.; Ding, H.; Dong, Q.; Chen, X.; Cui, M. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2018, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, T.; Iwata, M.; Shibushita, M.; Tsunetomi, K.; Nagata, M.; Kajitani, N.; Miura, A.; Matsuo, R.; Nishiguchi, T.; Kato, T.A.; et al. Beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, attenuates anxiety-related behavior in a rodent post-traumatic stress disorder model. Sci. Rep. 2020, 10, 21629. [Google Scholar] [CrossRef]
- Polito, R.; La Torre, M.E.; Moscatelli, F.; Cibelli, G.; Valenzano, A.; Panaro, M.A.; Monda, M.; Messina, A.; Monda, V.; Pisanelli, D.; et al. The Ketogenic Diet and Neuroinflammation: The Action of Beta-Hydroxybutyrate in a Microglial Cell Line. Int. J. Mol. Sci. 2023, 24, 3102. [Google Scholar] [CrossRef]
- Jin, L.W.; Di Lucente, J.; Ruiz Mendiola, U.; Suthprasertporn, N.; Tomilov, A.; Cortopassi, G.; Kim, K.; Ramsey, J.J.; Maezawa, I. The ketone body beta-hydroxybutyrate shifts microglial metabolism and suppresses amyloid-beta oligomer-induced inflammation in human microglia. FASEB J. 2023, 37, e23261. [Google Scholar] [CrossRef]
- Huang, C.; Wang, P.; Xu, X.; Zhang, Y.; Gong, Y.; Hu, W.; Gao, M.; Wu, Y.; Ling, Y.; Zhao, X.; et al. The ketone body metabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 2018, 66, 256–278. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Torres-Esquivel, C.; Montiel, T.; Flores-Mendez, M.; Massieu, L. Effect of beta-Hydroxybutyrate on Autophagy Dynamics During Severe Hypoglycemia and the Hypoglycemic Coma. Front. Cell Neurosci. 2020, 14, 547215. [Google Scholar] [CrossRef] [PubMed]
- Camberos-Luna, L.; Geronimo-Olvera, C.; Montiel, T.; Rincon-Heredia, R.; Massieu, L. The Ketone Body, beta-Hydroxybutyrate Stimulates the Autophagic Flux and Prevents Neuronal Death Induced by Glucose Deprivation in Cortical Cultured Neurons. Neurochem. Res. 2016, 41, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.F.; Nguyen, N.T.K.; Hsia, S.M.; Chen, H.Y.; Lin, S.H.; Lin, C.I. Protective Potential of beta-Hydroxybutyrate against Glucose-Deprivation-Induced Neurotoxicity Involving the Modulation of Autophagic Flux and the Monomeric Abeta Level in Neuro-2a Cells. Biomedicines 2023, 11, 698. [Google Scholar] [CrossRef]
- Montiel, T.; Gomora-Garcia, J.C.; Geronimo-Olvera, C.; Heras-Romero, Y.; Bernal-Vicente, B.N.; Perez-Martinez, X.; Tovar, Y.R.L.B.; Massieu, L. Modulation of the autophagy-lysosomal pathway and endoplasmic reticulum stress by ketone bodies in experimental models of stroke. J. Neurochem. 2023, 166, 87–106. [Google Scholar] [CrossRef]
- Liskiewicz, D.; Liskiewicz, A.; Nowacka-Chmielewska, M.M.; Grabowski, M.; Pondel, N.; Grabowska, K.; Student, S.; Barski, J.J.; Malecki, A. Differential Response of Hippocampal and Cerebrocortical Autophagy and Ketone Body Metabolism to the Ketogenic Diet. Front. Cell Neurosci. 2021, 15, 733607. [Google Scholar] [CrossRef]
- Hu, L.T.; Xie, X.Y.; Zhou, G.F.; Wen, Q.X.; Song, L.; Luo, B.; Deng, X.J.; Pan, Q.L.; Chen, G.J. HMGCS2-Induced Autophagic Degradation of Tau Involves Ketone Body and ANKRD24. J. Alzheimers Dis. 2023, 91, 407–426. [Google Scholar] [CrossRef]
- Hu, L.T.; Zhu, B.L.; Lai, Y.J.; Long, Y.; Zha, J.S.; Hu, X.T.; Zhang, J.H.; Chen, G.J. HMGCS2 promotes autophagic degradation of the amyloid-beta precursor protein through ketone body-mediated mechanisms. Biochem. Biophys. Res. Commun. 2017, 486, 492–498. [Google Scholar] [CrossRef]
- Wang, B.H.; Hou, Q.; Lu, Y.Q.; Jia, M.M.; Qiu, T.; Wang, X.H.; Zhang, Z.X.; Jiang, Y. Ketogenic diet attenuates neuronal injury via autophagy and mitochondrial pathways in pentylenetetrazol-kindled seizures. Brain Res. 2018, 1678, 106–115. [Google Scholar] [CrossRef]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Jana, M.; Modi, K.; Gonzalez, F.J.; Sims, K.B.; Berry-Kravis, E.; Pahan, K. Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: Implications for lysosomal storage disorders. J. Biol. Chem. 2015, 290, 10309–10324. [Google Scholar] [CrossRef] [PubMed]
- Montiel, T.; Montes-Ortega, L.A.; Flores-Yanez, S.; Massieu, L. Treatment with the Ketone Body D-beta-hydroxybutyrate Attenuates Autophagy Activated by NMDA and Reduces Excitotoxic Neuronal Damage in the Rat Striatum In Vivo. Curr. Pharm. Des. 2020, 26, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rodriguez, D.; Gimenez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.M.; Risa, O.; Sonnewald, U.; Schousboe, A.; Waagepetersen, H.S. Availability of neurotransmitter glutamate is diminished when beta-hydroxybutyrate replaces glucose in cultured neurons. J. Neurochem. 2009, 110, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Zilberter, M.; Ivanov, A.; Ziyatdinova, S.; Mukhtarov, M.; Malkov, A.; Alpar, A.; Tortoriello, G.; Botting, C.H.; Fulop, L.; Osypov, A.A.; et al. Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer’s disease. J. Neurochem. 2013, 125, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef]
- Lund, T.M.; Ploug, K.B.; Iversen, A.; Jensen, A.A.; Jansen-Olesen, I. The metabolic impact of beta-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J. Neurochem. 2015, 132, 520–531. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Pflanz, N.C.; Daszkowski, A.W.; James, K.A.; Mihic, S.J. Ketone body modulation of ligand-gated ion channels. Neuropharmacology 2019, 148, 21–30. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Won, Y.J.; Lu, V.B.; Puhl, H.L., 3rd; Ikeda, S.R. beta-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J. Neurosci. 2013, 33, 19314–19325. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, L.; Schröder, J.; Schuster, I.S.; Nakai, M.; Sun, G.; Sun, Y.B.Y.; Marino, E.; Degli-Esposti, M.A.; Marques, F.Z.; et al. Dietary fiber and microbiota metabolite receptors enhance cognition and alleviate disease in the 5xFAD mouse model of Alzheimer’s disease. J. Neurosci. 2023, 43, 6460–6475. [Google Scholar] [CrossRef]
- Rawat, K.; Singh, N.; Kumari, P.; Saha, L. A review on preventive role of ketogenic diet (KD) in CNS disorders from the gut microbiota perspective. Rev. Neurosci. 2021, 32, 143–157. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. The Role of Gut Microbiota in an Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 915. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, M.; Qian, J.; Wang, C.; Zhang, J. The Bridge Between Ischemic Stroke and Gut Microbes: Short-Chain Fatty Acids. Cell Mol. Neurobiol. 2023, 43, 543–559. [Google Scholar] [CrossRef]
- Attaye, I.; van Oppenraaij, S.; Warmbrunn, M.V.; Nieuwdorp, M. The Role of the Gut Microbiota on the Beneficial Effects of Ketogenic Diets. Nutrients 2021, 14, 191. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.t.; Mazmanian, S.K. The gut microbiota-brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Mulero, A.; Tinahones, A.; Bandera, B.; Moreno-Indias, I.; Macias-Gonzalez, M.; Tinahones, F.J. Keto microbiota: A powerful contributor to host disease recovery. Rev. Endocr. Metab. Disord. 2019, 20, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Krakovski, M.A.; Arora, N.; Jain, S.; Glover, J.; Dombrowski, K.; Hernandez, B.; Yadav, H.; Sarma, A.K. Diet-microbiome-gut-brain nexus in acute and chronic brain injury. Front. Neurosci. 2022, 16, 1002266. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, H.J.; Nagpal, R.; Yadav, H. Diet-Microbiota-Brain Axis in Alzheimer’s Disease. Ann. Nutr. Metab. 2021, 77 (Suppl. S2), 21–27. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.M.; Letchumanan, V.; Tan, L.T.; Hong, K.W.; Wong, S.H.; Ab Mutalib, N.S.; Lee, L.H.; Law, J.W. Ketogenic Diet: A Dietary Intervention via Gut Microbiome Modulation for the Treatment of Neurological and Nutritional Disorders (a Narrative Review). Nutrients 2022, 14, 3566. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Q.; Liu, J. Microbiota-gut-brain axis: A novel potential target of ketogenic diet for epilepsy. Curr. Opin. Pharmacol. 2021, 61, 36–41. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275.e16. [Google Scholar] [CrossRef] [PubMed]
- Newell, C.; Bomhof, M.R.; Reimer, R.A.; Hittel, D.S.; Rho, J.M.; Shearer, J. Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism. 2016, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Olivito, I.; Avolio, E.; Minervini, D.; Soda, T.; Rocca, C.; Angelone, T.; Iaquinta, F.S.; Bellizzi, D.; De Rango, F.; Bruno, R.; et al. Ketogenic diet ameliorates autism spectrum disorders-like behaviors via reduced inflammatory factors and microbiota remodeling in BTBR T(+) Itpr3(tf)/J mice. Exp. Neurol. 2023, 366, 114432. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, X.; Zhang, H.; Yin, J.; Zhao, P.; Yin, Q.; Wang, Z. Ketogenic diet protects MPTP-induced mouse model of Parkinson’s disease via altering gut microbiota and metabolites. MedComm 2023, 4, e268. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, S.; Huang, X.; Tong, H.; Niu, H.; Lu, L. Neuroprotective effect of a medium-chain triglyceride ketogenic diet on MPTP-induced Parkinson’s disease mice: A combination of transcriptomics and metabolomics in the substantia nigra and fecal microbiome. Cell Death Discov. 2023, 9, 251. [Google Scholar] [CrossRef]
- Swidsinski, A.; Dorffel, Y.; Loening-Baucke, V.; Gille, C.; Goktas, O.; Reisshauer, A.; Neuhaus, J.; Weylandt, K.H.; Guschin, A.; Bock, M. Reduced Mass and Diversity of the Colonic Microbiome in Patients with Multiple Sclerosis and Their Improvement with Ketogenic Diet. Front. Microbiol. 2017, 8, 1141. [Google Scholar] [CrossRef]
- Park, S.M.; Zhang, T.; Wu, X.G.; Qiu, J.Y. Ketone production by ketogenic diet and by intermittent fasting has different effects on the gut microbiota and disease progression in an Alzheimer’s disease rat model. J. Clin. Biochem. Nutr. 2020, 67, 188–198. [Google Scholar] [CrossRef]
- Miyamoto, J.; Ohue-Kitano, R.; Mukouyama, H.; Nishida, A.; Watanabe, K.; Igarashi, M.; Irie, J.; Tsujimoto, G.; Satoh-Asahara, N.; Itoh, H.; et al. Ketone body receptor GPR43 regulates lipid metabolism under ketogenic conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 23813–23821. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Population | Number of Participants | Trial Type | Intervention/ Therapy | Duration | Outcome | Reference |
|---|---|---|---|---|---|---|
| MCI | 23 | RCT parallel | KD | 6 weeks | Improved memory function | [50] |
| MCI | 6 | RCT parallel | 56 g/day MCTG | 24 weeks | Improved memory | [51] |
| MCI | 20 | RCT cross over | Modified Mediterranean KD | 6 weeks | Improved memory performance, brain metabolism | [52] |
| MCI | 83 | RCT parallel | 15 g twice/day MCTG drink | 6 months | Improved episodic memory, language, executive function | [53] |
| AD, MCI | 20 | RCT cross over | 40 mL MCTG drink | 1 dose (single test day | Improved cognitive scores in ApoE4(-) subjects | [54] |
| AD | 152 | RCT parallel | MCTG (AC-1202) | 90 days | Improved cognitive performance in ApoE4(-) subjects | [55] |
| AD | 1 | Single arm | MCTG or ketone ester | 20 months | Improved mood, cognitive, and daily activity | [56] |
| AD | 15 | Single arm | MCTG supplemented KD | 3 months | Improved cognitive scores | [57] |
| AD | 20 | Single arm | 20 g/day MCTG ketogenic meals | 12 weeks | Improved logical memory | [58] |
| AD | 49 | RCT cross over | 17.3 g/day MCTG | 4 weeks | Improved cognition in ApoE4(-) subjects | [59] |
| AD | 26 | RCT cross over | KD | 12 weeks | Improved daily function, quality of life | [60] |
| PD | 7 | Single arm | KD | 28 days | Improved motor symptoms | [61] |
| PD | 47 | RCT parallel | KD | 8 weeks | Improved motor and non-motor symptoms | [62] |
| PD | 14 | RCT parallel | Low-carb KD | 8 weeks | Improved vocabulary and memory | [63] |
| MS | 60 | RCT parallel | KD | 3–6 months | Improved quality of life and mental health | [64] |
| MS | 20 | Single arm | Modified Atkins diet KD | 6 months | Improved fatigue and depression scores | [30] |
| HD | 1 | Single arm | Time-restricted KD | 48 weeks | Improved motor symptoms, HD-related behavior problems, quality of life | [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.; Kim, S.R.; Lee, J.E.; Lee, S.; Son, H.J.; Choe, W.; Yoon, K.-S.; Kim, S.S.; Yeo, E.-J.; Kang, I. Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 124. https://doi.org/10.3390/ijms25010124
Jang J, Kim SR, Lee JE, Lee S, Son HJ, Choe W, Yoon K-S, Kim SS, Yeo E-J, Kang I. Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2024; 25(1):124. https://doi.org/10.3390/ijms25010124
Chicago/Turabian StyleJang, Jiwon, Su Rim Kim, Jo Eun Lee, Seoyeon Lee, Hyeong Jig Son, Wonchae Choe, Kyung-Sik Yoon, Sung Soo Kim, Eui-Ju Yeo, and Insug Kang. 2024. "Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases" International Journal of Molecular Sciences 25, no. 1: 124. https://doi.org/10.3390/ijms25010124
APA StyleJang, J., Kim, S. R., Lee, J. E., Lee, S., Son, H. J., Choe, W., Yoon, K.-S., Kim, S. S., Yeo, E.-J., & Kang, I. (2024). Molecular Mechanisms of Neuroprotection by Ketone Bodies and Ketogenic Diet in Cerebral Ischemia and Neurodegenerative Diseases. International Journal of Molecular Sciences, 25(1), 124. https://doi.org/10.3390/ijms25010124