
Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
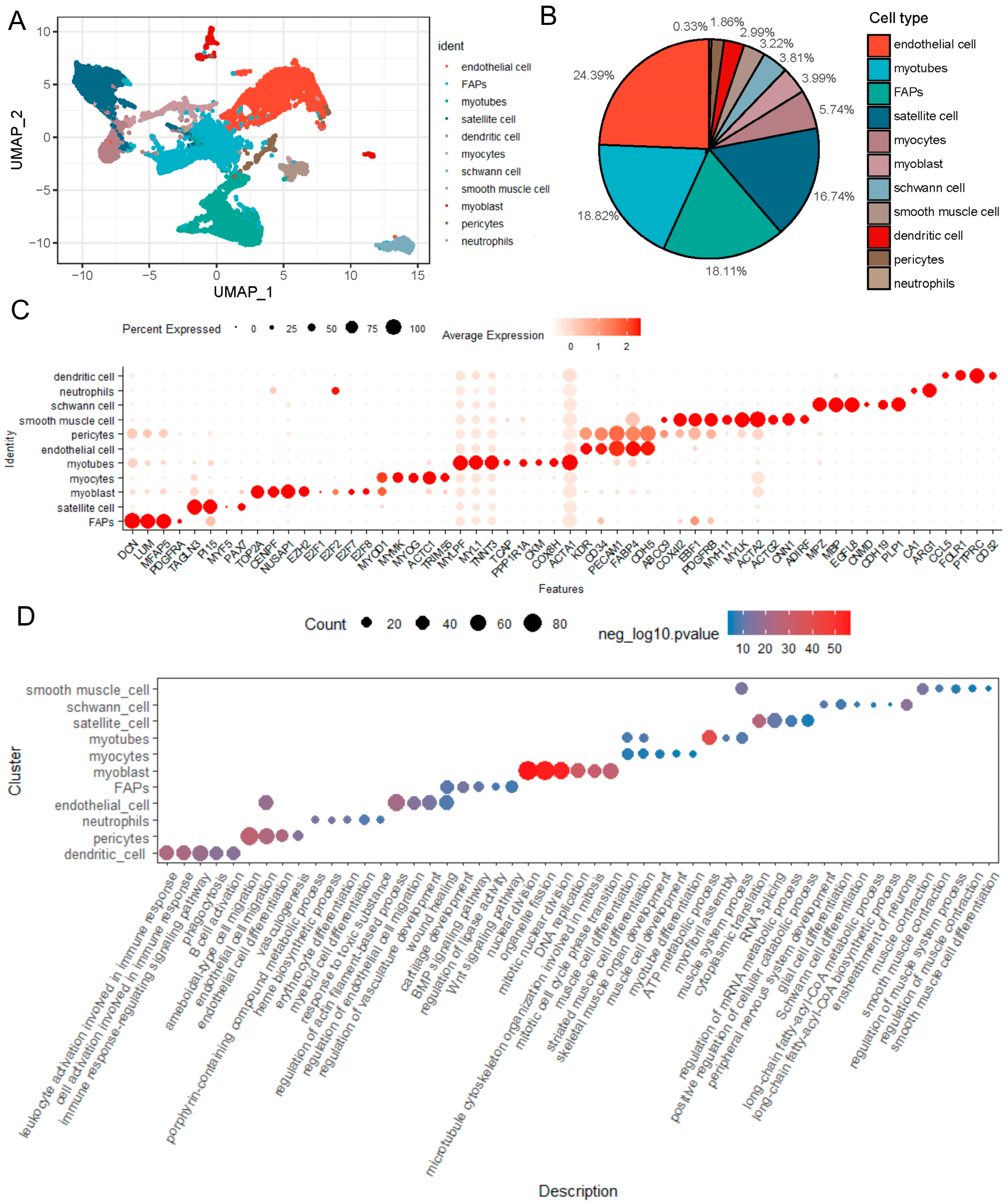
2.1. Identification the Cell Populations of LD of a 1-Day-Old Newborn Suhuai Boar
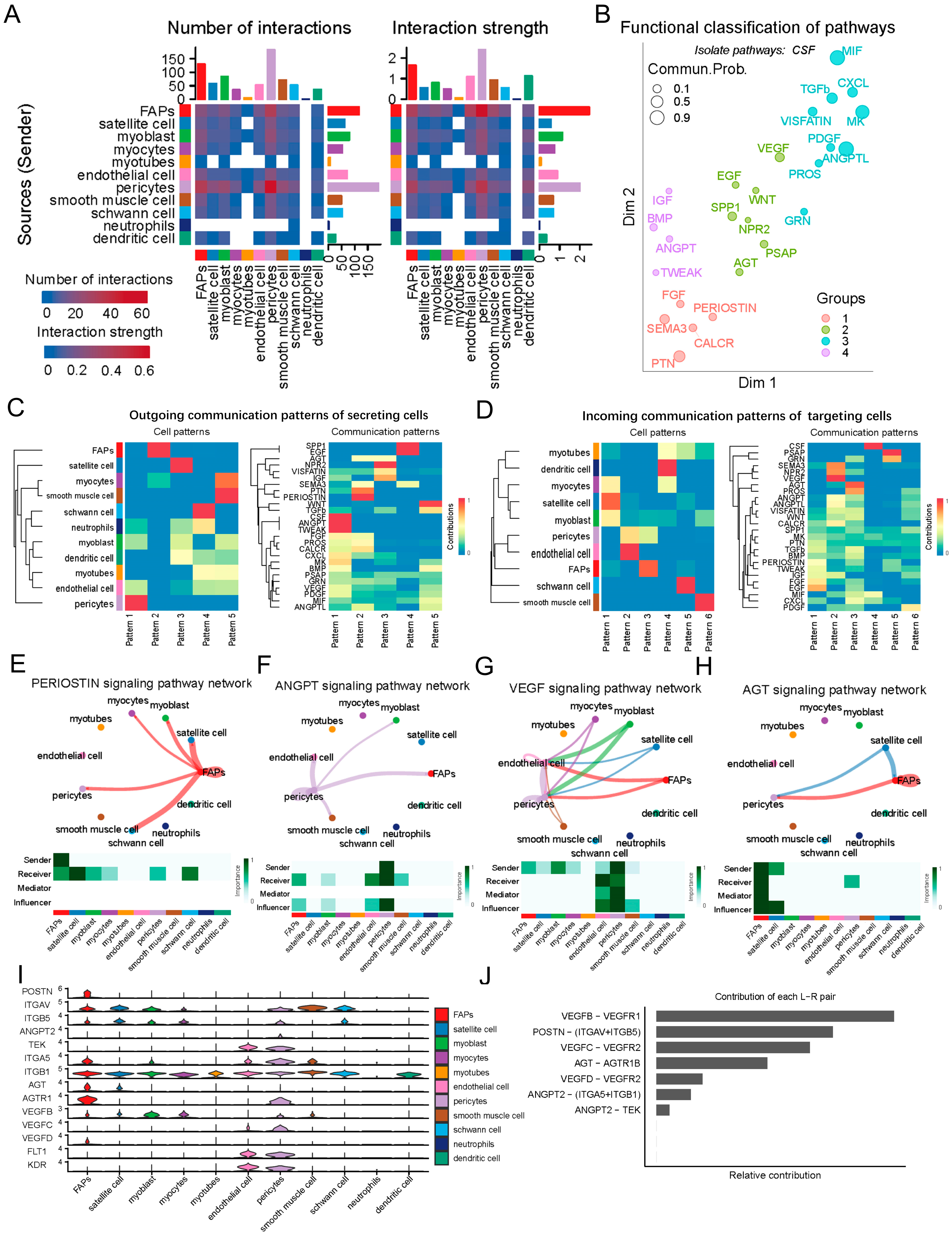
2.2. Analysis of Cell Communication in the LD of the Newborn Suhuai Pig by CellChat
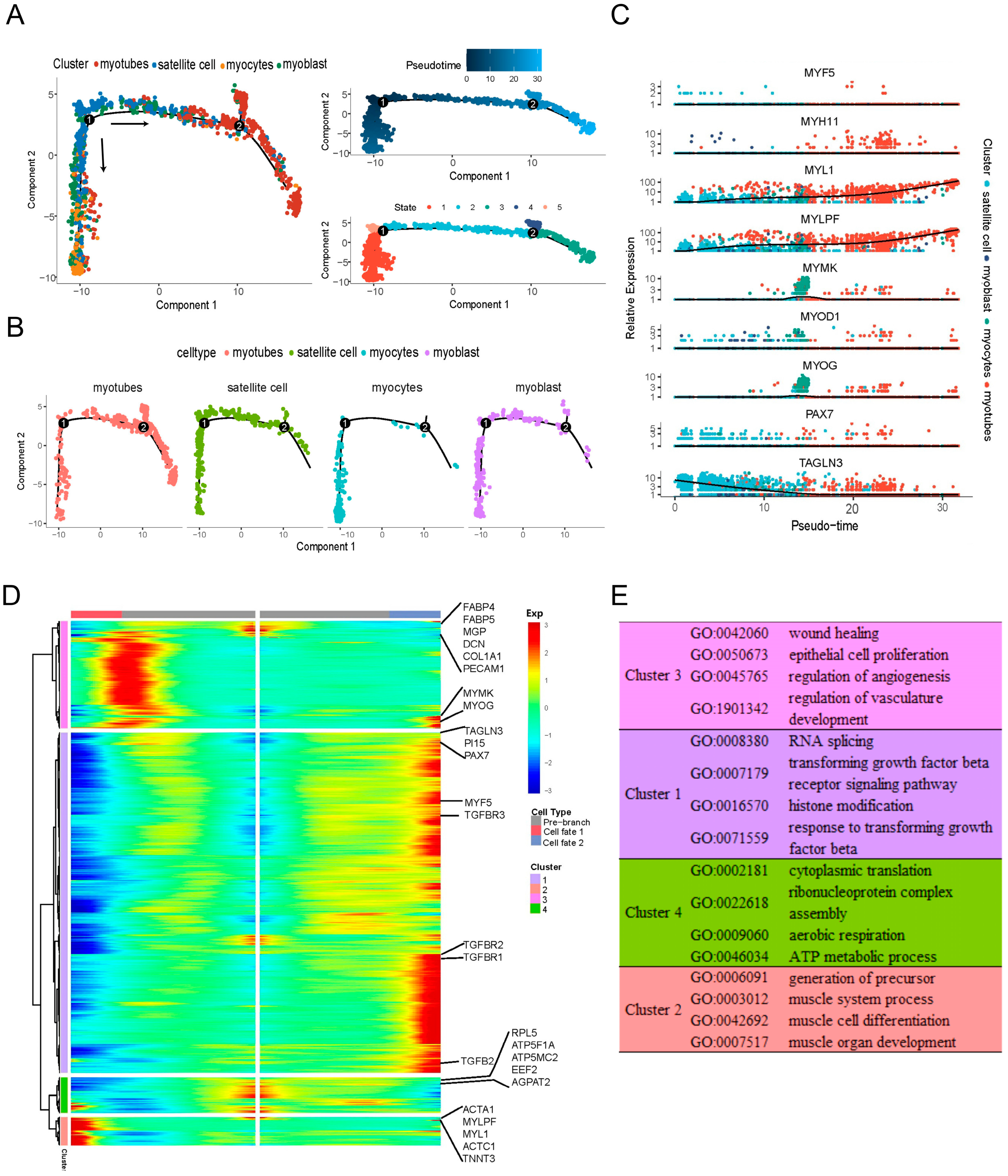
2.3. Pseudotime Patterns Reveal the Relationships among Different Myogenic Populations
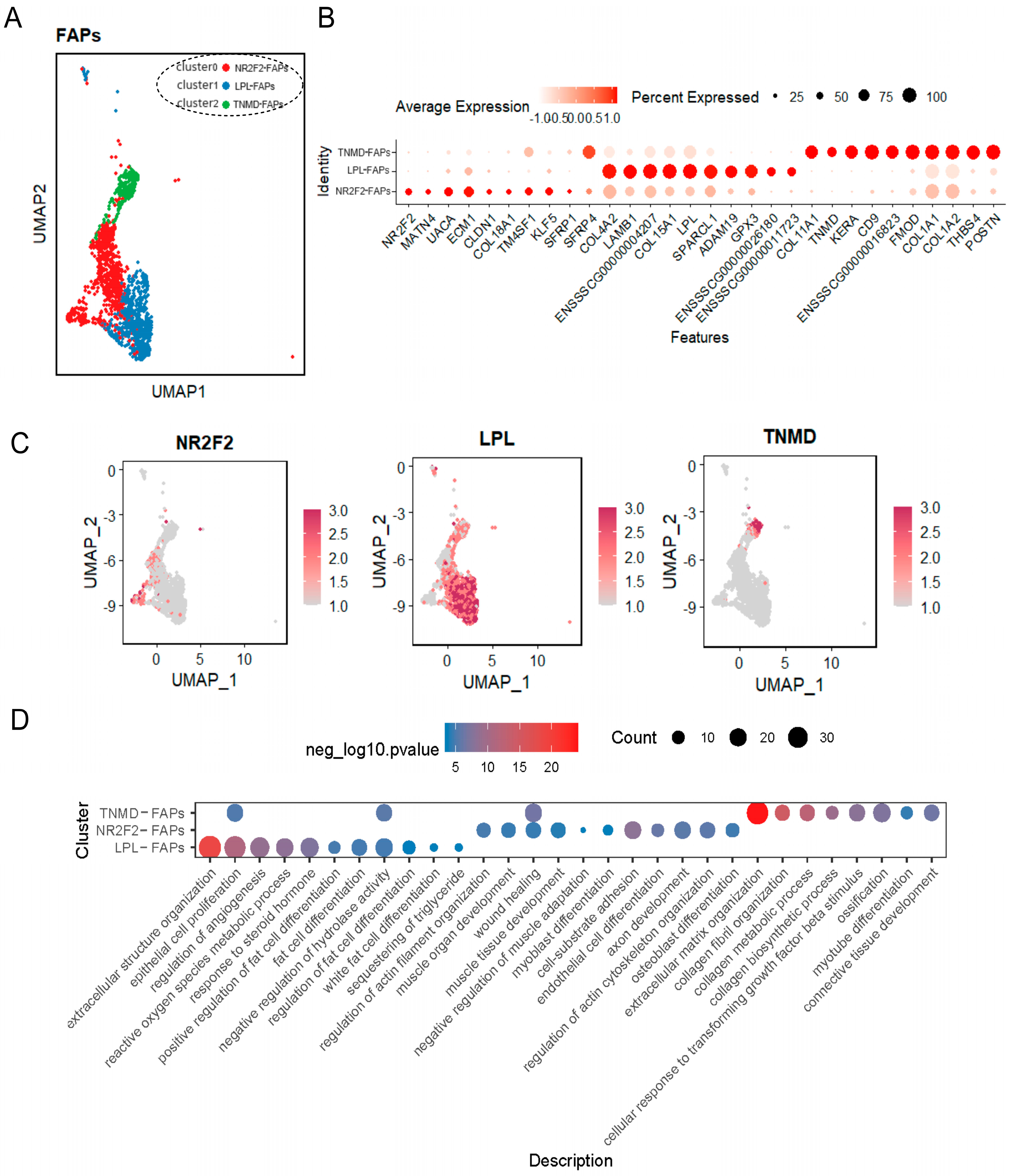
2.4. FAPs Were Associated with the Differentiation of Tendon
3. Discussion
4. Materials and Methods
4.1. Animal and Sample Collection
4.2. Preparation of Single-Cell Suspensions
4.3. Generating the 10× Genomics ScRNA-Seq Library and Sequencing
4.4. Sequencing Data Quality Control
4.5. Cell Clustering and Cell Type Annotation
4.6. Analysis of Cellular Communication Networks
4.7. Trajectory Analysis of Myogenic Cell
4.8. Gene Ontology (GO) Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Long, Y.; Zhang, Y.F.; Zhang, Z.Y.; Yang, B.; Chen, C.Y.; Huang, L.S.; Su, Y. Phenotypic and genetic correlations of pork myoglobin content with meat colour and other traits in an eight breed-crossed heterogeneous population. Anim. Int. J. Anim. Biosci. 2021, 15, 100364. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Li, A.; Xie, L.; Miao, X. Genome-Wide Analysis of mRNAs and lncRNAs of Intramuscular Fat Related to Lipid Metabolism in Two Pig Breeds. Cell. Physiol. Biochem. 2018, 50, 2406–2422. [Google Scholar] [CrossRef]
- Jang, D.; Yoon, J.; Taye, M.; Lee, W.; Kwon, T.; Shim, S.; Kim, H. Multivariate genome-wide association studies on tenderness of Berkshire and Duroc pig breeds. Genes Genom. 2018, 40, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Cai, S.; Hu, B.; Wang, X.; Liu, T.; Lin, Z.; Tong, X.; Xu, R.; Chen, M.; Duo, T.; Zhu, Q.; et al. Integrative single-cell RNA-seq and ATAC-seq analysis of myogenic differentiation in pig. BMC Biol. 2023, 21, 19. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wan, B.; Qiu, K.; Wang, Y.; Zhang, X.; Jiao, N.; Yan, E.; Wu, J.; Yu, R.; Gao, S.; et al. Single-Cell RNA-Sequencing Provides Insight into Skeletal Muscle Evolution during the Selection of Muscle Characteristics. Adv. Sci. 2023, 10, e2305080. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tang, Q.; Hu, S.; Chen, Z.; Zhou, X.; Zeng, B.; Wang, Y.; He, M.; Li, Y.; Gui, L.; et al. A pig BodyMap transcriptome reveals diverse tissue physiologies and evolutionary dynamics of transcription. Nat. Commun. 2021, 12, 3715. [Google Scholar] [CrossRef]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Jones, R.C.; Karkanias, J.; Krasnow, M.A.; Pisco, A.O.; Quake, S.R.; Salzman, J.; Yosef, N.; Bulthaup, B.; Brown, P.; Harper, W.; et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376, eabl4896. [Google Scholar] [CrossRef]
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Crow, M.; Rice, B.R.; Li, F.; Harris, B.; Liu, L.; Demesa-Arevalo, E.; Lu, Z.; Wang, L.; Fox, N.; et al. Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell 2021, 56, 557–568.e556. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Xiong, L.; Guo, S.; Wang, X.; La, Y.; Chu, M.; Liang, C.; Yan, P.; Guo, X. Single-Cell Transcriptomics Analysis Reveals a Cell Atlas and Cell Communication in Yak Ovary. Int. J. Mol. Sci. 2023, 24, 1839. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Minne, M.; Eekhout, T.; De Rybel, B. Single Cell RNA-Sequencing in Arabidopsis Root Tissues. Methods Mol. Biol. 2023, 2698, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jara, C.P.; Wang, O.; Shi, Y.; Wu, X.; Thibivilliers, S.; Wóycicki, R.K.; Carlson, M.A.; Velander, W.H.; Araújo, E.P.; et al. Isolating and cryopreserving pig skin cells for single-cell RNA sequencing study. PLoS ONE 2022, 17, e0263869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, J.; Wang, H.; Xia, J.; Liu, P.; Chen, F.; Jiang, H.; Miao, Q.; Wu, W.; Zhang, L.; et al. A high-resolution cell atlas of the domestic pig lung and an online platform for exploring lung single-cell data. J. Genet. Genom. Yi Chuan Xue Bao 2021, 48, 411–425. [Google Scholar] [CrossRef]
- Qiu, K.; Xu, D.; Wang, L.; Zhang, X.; Jiao, N.; Gong, L.; Yin, J. Association Analysis of Single-Cell RNA Sequencing and Proteomics Reveals a Vital Role of Ca2+ Signaling in the Determination of Skeletal Muscle Development Potential. Cells 2020, 9, 1045. [Google Scholar] [CrossRef]
- Herrera-Uribe, J.; Wiarda, J.E.; Sivasankaran, S.K.; Daharsh, L.; Liu, H.; Byrne, K.A.; Smith, T.P.L.; Lunney, J.K.; Loving, C.L.; Tuggle, C.K. Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing. Front. Genet. 2021, 12, 689406. [Google Scholar] [CrossRef]
- Wang, F.; Ding, P.; Liang, X.; Ding, X.; Brandt, C.B.; Sjöstedt, E.; Zhu, J.; Bolund, S.; Zhang, L.; de Rooij, L.; et al. Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level. Nat. Commun. 2022, 13, 3620. [Google Scholar] [CrossRef]
- Wang, B.; Li, P.; Hou, L.; Zhou, W.; Tao, W.; Liu, C.; Liu, K.; Niu, P.; Zhang, Z.; Li, Q.; et al. Genome-wide association study and genomic prediction for intramuscular fat content in Suhuai pigs using imputed whole-genome sequencing data. Evol. Appl. 2022, 15, 2054–2066. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Giuliani, G.; Rosina, M.; Reggio, A. Signaling pathways regulating the fate of fibro/adipogenic progenitors (FAPs) in skeletal muscle regeneration and disease. FEBS J. 2022, 289, 6484–6517. [Google Scholar] [CrossRef]
- Hakanpaa, L.; Sipila, T.; Leppanen, V.M.; Gautam, P.; Nurmi, H.; Jacquemet, G.; Eklund, L.; Ivaska, J.; Alitalo, K.; Saharinen, P. Endothelial destabilization by angiopoietin-2 via integrin β1 activation. Nat. Commun. 2015, 6, 5962. [Google Scholar] [CrossRef]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Lu, H.; Cassis, L.A.; Kooi, C.W.; Daugherty, A. Structure and functions of angiotensinogen. Hypertens. Res. 2016, 39, 492–500. [Google Scholar] [CrossRef]
- Pahlavani, M.; Kalupahana, N.S.; Ramalingam, L.; Moustaid-Moussa, N. Regulation and Functions of the Renin-Angiotensin System in White and Brown Adipose Tissue. Compr. Physiol. 2017, 7, 1137–1150. [Google Scholar] [CrossRef]
- Durheim, M.T.; Slentz, C.A.; Bateman, L.A.; Mabe, S.K.; Kraus, W.E. Relationships between exercise-induced reductions in thigh intermuscular adipose tissue, changes in lipoprotein particle size, and visceral adiposity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E407–E412. [Google Scholar] [CrossRef]
- Xu, Z.; You, W.; Chen, W.; Zhou, Y.; Nong, Q.; Valencak, T.G.; Wang, Y.; Shan, T. Single-cell RNA sequencing and lipidomics reveal cell and lipid dynamics of fat infiltration in skeletal muscle. J. Cachexia Sarcopenia Muscle 2021, 12, 109–129. [Google Scholar] [CrossRef]
- Li, X.; Fu, X.; Yang, G.; Du, M. Review: Enhancing intramuscular fat development via targeting fibro-adipogenic progenitor cells in meat animals. Anim. Int. J. Anim. Biosci. 2020, 14, 312–321. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kardon, G. Muscle and tendon morphogenesis in the avian hind limb. Development 1998, 125, 4019–4032. [Google Scholar] [CrossRef]
- Ros, M.A.; Rivero, F.B.; Hinchliffe, J.R.; Hurle, J.M. Immunohistological and ultrastructural study of the developing tendons of the avian foot. Anat. Embryol. 1995, 192, 483–496. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sakiyama, K.; Kitamura, K.; Yamamoto, Y.; Takagi, T.; Sekiya, S.; Watanabe, G.; Taniguchi, S.; Ogawa, Y.; Ishizuka, S.; et al. Development and Regeneration of Muscle, Tendon, and Myotendinous Junctions in Striated Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 3006. [Google Scholar] [CrossRef]
- Reggio, A.; Rosina, M.; Palma, A.; Cerquone Perpetuini, A.; Petrilli, L.L.; Gargioli, C.; Fuoco, C.; Micarelli, E.; Giuliani, G.; Cerretani, M.; et al. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/β-catenin axis. Cell Death Differ. 2020, 27, 2921–2941. [Google Scholar] [CrossRef]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Chen, Z.; Li, X.Y.; Guo, P.; Wang, D.L. MYBPC2 and MYL1 as Significant Gene Markers for Rhabdomyosarcoma. Technol. Cancer Res. Treat. 2021, 20, 1533033820979669. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e720. [Google Scholar] [CrossRef] [PubMed]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise review: Evidence for CD34 as a common marker for diverse progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhang, R.; Kuang, X.; Yu, C.; Niu, S.; Du, Y.; Lu, D.; Li, S.; Teng, Z.; Lu, S. Single-cell RNA-seq reveals interferon-induced guanylate-binding proteins are linked with sarcopenia. J. Cachexia Sarcopenia Muscle 2022, 13, 2985–2998. [Google Scholar] [CrossRef]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef]
- Lu, G.; Du, R.; Liu, Y.; Zhang, S.; Li, J.; Pei, J. RGS5 as a Biomarker of Pericytes, Involvement in Vascular Remodeling and Pulmonary Arterial Hypertension. Vasc. Health Risk Manag. 2023, 19, 673–688. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Ohtani, K.; DeGregori, J.; Leone, G.; Herendeen, D.R.; Kelly, T.J.; Nevins, J.R. Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol. Cell. Biol. 1996, 16, 6977–6984. [Google Scholar] [CrossRef]
- Yan, Z.; Choi, S.; Liu, X.; Zhang, M.; Schageman, J.J.; Lee, S.Y.; Hart, R.; Lin, L.; Thurmond, F.A.; Williams, R.S. Highly coordinated gene regulation in mouse skeletal muscle regeneration. J. Biol. Chem. 2003, 278, 8826–8836. [Google Scholar] [CrossRef]
- Parakati, R.; Dimario, J.X. Repression of fibroblast growth factor receptor 1 gene expression by E2F4 in skeletal muscle cells. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 232, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Rudar, M.; Fiorotto, M.L.; Davis, T.A. Regulation of Muscle Growth in Early Postnatal Life in a Swine Model. Annu. Rev. Anim. Biosci. 2019, 7, 309–335. [Google Scholar] [CrossRef] [PubMed]
- Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110. [Google Scholar] [CrossRef] [PubMed]
- Salingcarnboriboon, R.; Yoshitake, H.; Tsuji, K.; Obinata, M.; Amagasa, T.; Nifuji, A.; Noda, M. Establishment of tendon-derived cell lines exhibiting pluripotent mesenchymal stem cell-like property. Exp. Cell Res. 2003, 287, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Docheva, D.; Hunziker, E.B.; Fässler, R.; Brandau, O. Tenomodulin is necessary for tenocyte proliferation and tendon maturation. Mol. Cell. Biol. 2005, 25, 699–705. [Google Scholar] [CrossRef]
- Kim, M.; Franke, V.; Brandt, B.; Lowenstein, E.D.; Schöwel, V.; Spuler, S.; Akalin, A.; Birchmeier, C. Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nat. Commun. 2020, 11, 6375. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, T.; Shukunami, C.; Amemiya-Kudo, M.; Shimano, H.; Ozaki, T. Scleraxis and E47 cooperatively regulate the Sox9-dependent transcription. Int. J. Biochem. Cell Biol. 2010, 42, 148–156. [Google Scholar] [CrossRef]
- Schweitzer, R.; Chyung, J.H.; Murtaugh, L.C.; Brent, A.E.; Rosen, V.; Olson, E.N.; Lassar, A.; Tabin, C.J. Analysis of the tendon cell fate using Scleraxis, a specific marker for tendons and ligaments. Development 2001, 128, 3855–3866. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, W.; Jiang, N.; Ji, Z.; Ni, M.; Zhang, Z.; Zhao, Q.; Huang, R.; Li, P.; Hou, L. Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig. Int. J. Mol. Sci. 2024, 25, 1204. https://doi.org/10.3390/ijms25021204
Xiao W, Jiang N, Ji Z, Ni M, Zhang Z, Zhao Q, Huang R, Li P, Hou L. Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig. International Journal of Molecular Sciences. 2024; 25(2):1204. https://doi.org/10.3390/ijms25021204
Chicago/Turabian StyleXiao, Wei, Nengjing Jiang, Zhengyu Ji, Mengru Ni, Zhaobo Zhang, Qingbo Zhao, Ruihua Huang, Pinghua Li, and Liming Hou. 2024. "Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig" International Journal of Molecular Sciences 25, no. 2: 1204. https://doi.org/10.3390/ijms25021204
APA StyleXiao, W., Jiang, N., Ji, Z., Ni, M., Zhang, Z., Zhao, Q., Huang, R., Li, P., & Hou, L. (2024). Single-Cell RNA Sequencing Reveals the Cellular Landscape of Longissimus Dorsi in a Newborn Suhuai Pig. International Journal of Molecular Sciences, 25(2), 1204. https://doi.org/10.3390/ijms25021204