

Probiotic-Induced Modulation of Microbiota Composition and Antibiotic Resistance Genes Load, an In Vitro Assessment

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Impact of Probiotics on Intestinal Microbiota Composition
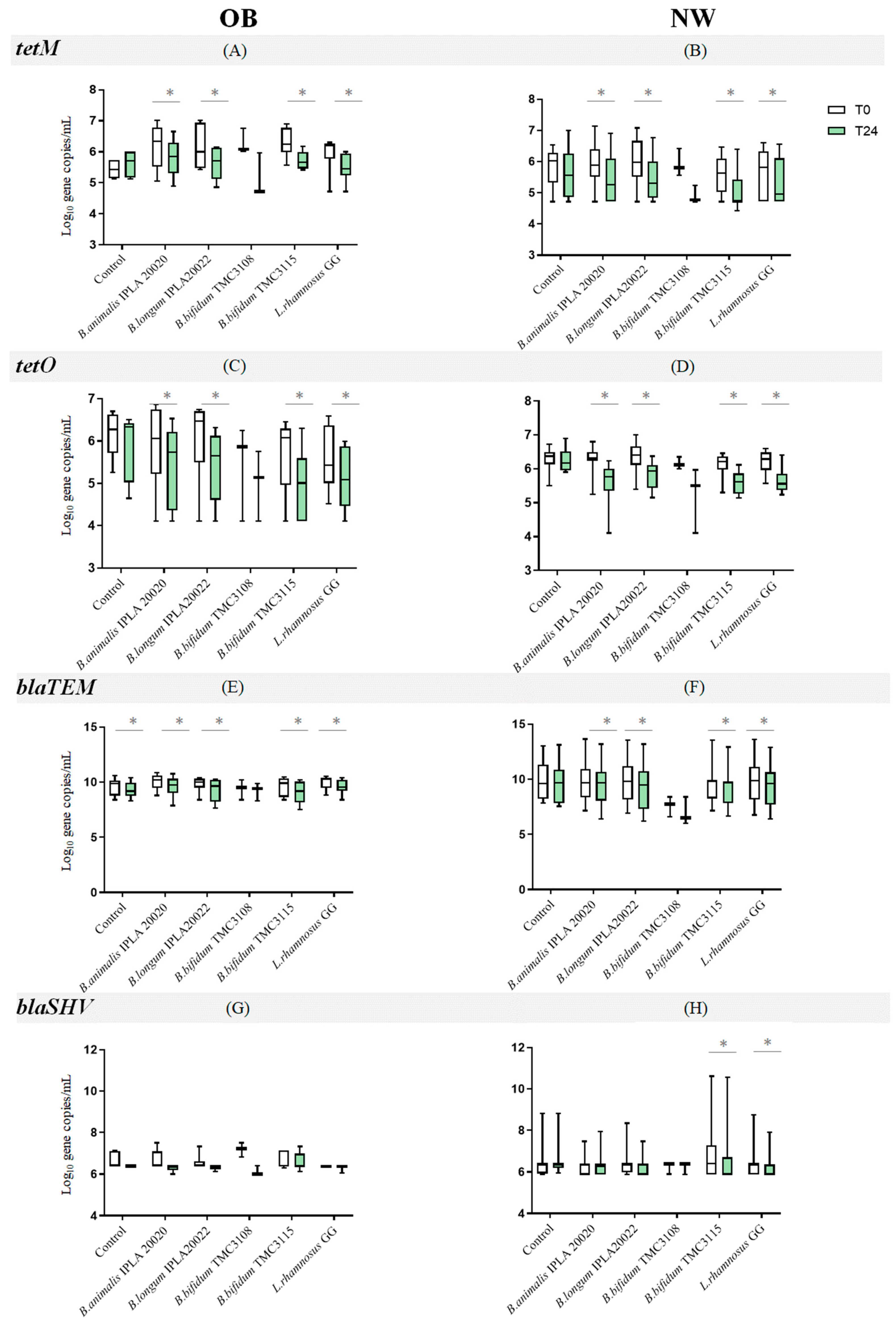
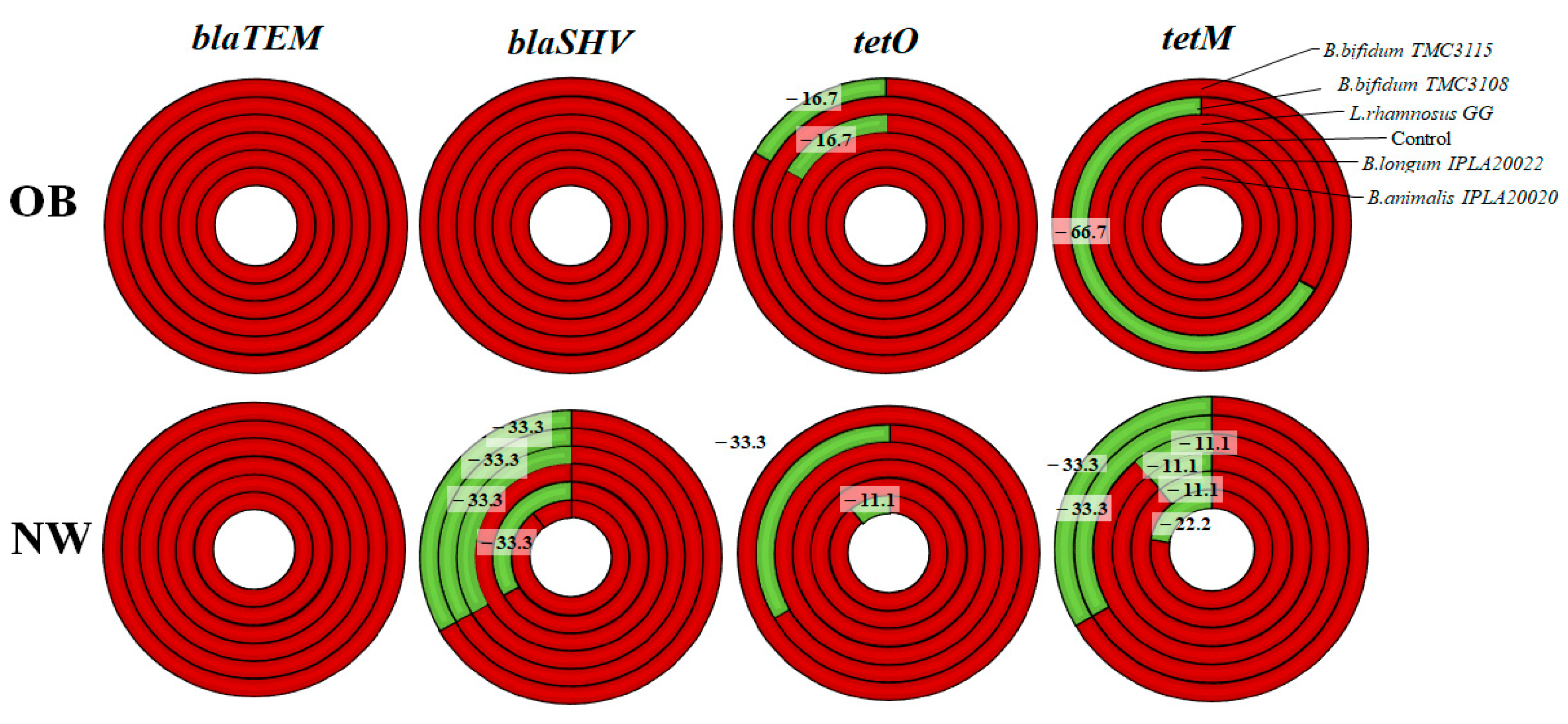
2.2. Impact of Probiotics on the Levels of Antibiotic Resistance Genes
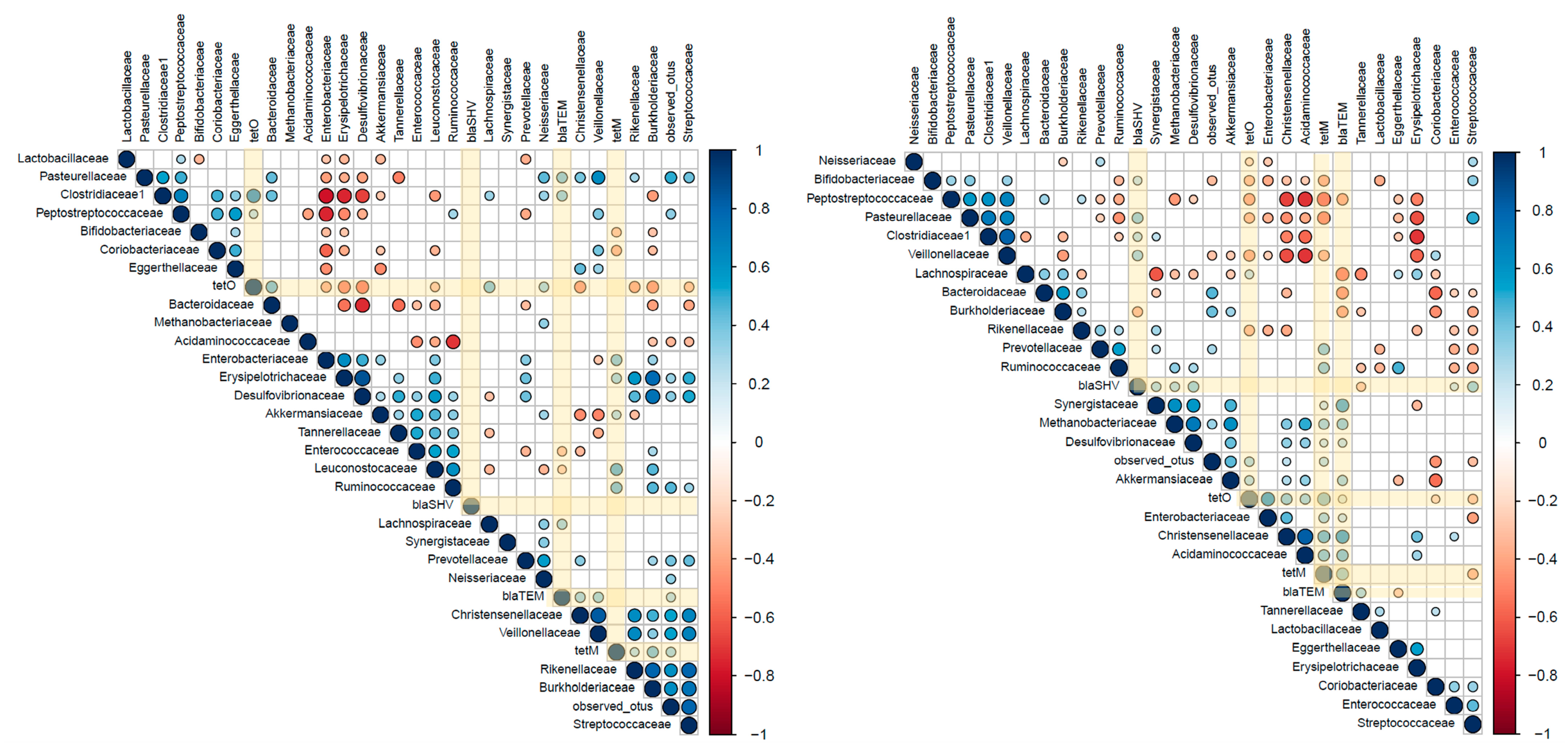
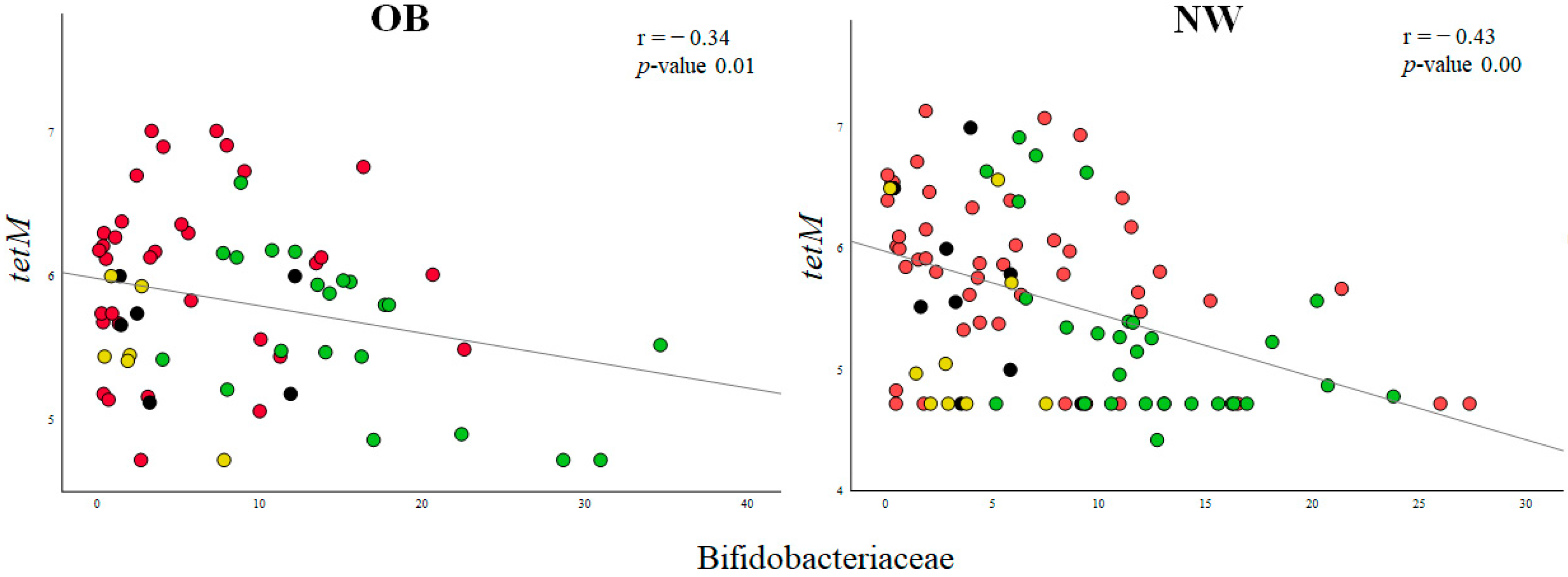
2.3. Associations between ARGs and Probiotic-Modulated Microbiota
3. Discussion
4. Materials and Methods
4.1. Fecal and Probiotic Culture Conditions
4.2. Microbiota Composition by Metataxonomic Analyses
4.3. Quantification of Antibiotic Resistance Genes
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-Analysis of Gut Microbiome Studies Identifies Disease-Specific and Shared Responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed]
- WHO Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 13 May 2020).
- Zhao, L. The Gut Microbiota and Obesity: From Correlation to Causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Duque, A.L.R.F.; Saad, S.M.I.; Sivieri, K. Gut Microbiome Approaches to Treat Obesity in Humans. Appl. Microbiol. Biotechnol. 2019, 103, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Role of Gut Microbiota in the Aetiology of Obesity: Proposed Mechanisms and Review of the Literature. J. Obes. 2016, 2016, 7353642. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms Underlying the Resistance to Diet-Induced Obesity in Germ-Free Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.J.; Magness, S.; Jobin, C.; Lund, P.K. High-Fat Diet: Bacteria Interactions Promote Intestinal Inflammation Which Precedes and Correlates with Obesity and Insulin Resistance in Mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Castaner, O.; Goday, A.; Park, Y.-M.; Lee, S.-H.; Magkos, F.; Shiow, S.-A.T.E.; Schröder, H. The Gut Microbiome Profile in Obesity: A Systematic Review. Int. J. Endocrinol. 2018, 2018, 4095789. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The Human Gut Microbiota: Metabolism and Perspective in Obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [PubMed]
- Finucane, M.M.; Sharpton, T.J.; Laurent, T.J.; Pollard, K.S. A Taxonomic Signature of Obesity in the Microbiome? Getting to the Guts of the Matter. PLoS ONE 2014, 9, e84689. [Google Scholar] [CrossRef]
- Sze, M.A.; Schloss, P.D. Looking for a Signal in the Noise: Revisiting Obesity and the Microbiome. mBio 2016, 7, e01018-16. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Xu, Z.; Knight, R. Meta-Analyses of Human Gut Microbes Associated with Obesity and IBD. FEBS Lett. 2014, 588, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.N.; Yao, Y.; Ju, S.Y. Short Chain Fatty Acids and Fecal Microbiota Abundance in Humans with Obesity: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 2512. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Tolone, S.; Gravina, A.G.; Patrone, V.; Romano, M.; Tuccillo, C.; Mozzillo, A.L.; Amoroso, V.; Misso, G.; et al. Gastrointestinal Hormones, Intestinal Microbiota and Metabolic Homeostasis in Obese Patients: Effect of Bariatric Surgery. In Vivo 2016, 30, 321–330. [Google Scholar]
- Magouliotis, D.E.; Tasiopoulou, V.S.; Sioka, E.; Chatedaki, C.; Zacharoulis, D. Impact of Bariatric Surgery on Metabolic and Gut Microbiota Profile: A Systematic Review and Meta-Analysis. Obes. Surg. 2017, 27, 1345–1357. [Google Scholar] [CrossRef]
- Nogacka, A.; los Reyes-Gavilán, C.G.; Martínez-Faedo, C.; Ruas-Madiedo, P.; Suarez, A.; Mancabelli, L.; Ventura, M.; Cifuentes, A.; León, C.; Gueimonde, M.; et al. Impact of Extreme Obesity and Diet-Induced Weight Loss on the Fecal Metabolome and Gut Microbiota. Mol. Nutr. Food Res. 2021, 65, 2000030. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazi, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major Microbiota Dysbiosis in Severe Obesity: Fate after Bariatric Surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Severe Obesity and Gut Microbiota: Does Bariatric Surgery Really Reset the System? Gut 2019, 68, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Borgeraas, H.; Johnson, L.K.; Skattebu, J.; Hertel, J.K.; Hjelmesæth, J. Effects of Probiotics on Body Weight, Body Mass Index, Fat Mass and Fat Percentage in Subjects with Overweight or Obesity: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Obes. Rev. 2018, 19, 219–232. [Google Scholar] [CrossRef]
- Ho, J.; Yeoh, Y.K.; Barua, N.; Chen, Z.; Lui, G.; Wong, S.H.; Yang, X.; Chan, M.C.; Chan, P.K.; Hawkey, P.M.; et al. Systematic Review of Human Gut Resistome Studies Revealed Variable Definitions and Approaches. Gut Microbes 2020, 12, 1700755. [Google Scholar] [CrossRef]
- EMA. European Medicine Agency. Available online: https://www.ema.europa.eu/en/documents/report/sales-veterinary-antimicrobial-agents-31-european-countries-2021-trends-2010-2021-twelfth-esvac_en.pdf (accessed on 15 October 2022).
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and Prebiotics in Intestinal Health and Disease: From Biology to the Clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. The International Scientific Association for Probiotics and Prebiotics Consensus Statement on the Scope and Appropriate Use of the Term Probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Sanders, M.E.; Merenstein, D.; Merrifield, C.A.; Hutkins, R. Probiotics for Human Use. Nutr. Bull. 2018, 43, 212–225. [Google Scholar] [CrossRef]
- Maldonado-Gómez, M.X.; Martínez, I.; Bottacini, F.; O’Callaghan, A.; Ventura, M.; van Sinderen, D.; Hillmann, B.; Vangay, P.; Knights, D.; Hutkins, R.W.; et al. Stable Engraftment of Bifidobacterium Longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe 2016, 20, 515–526. [Google Scholar] [CrossRef]
- FAO/WHO. Probiotics in Food. Health and Nutritional Proporties and Guidelines for Evaluation; FAO/WHO: Rome, Italy, 2006; ISBN 92-5-105513-0.
- Binda, S.; Hill, C.; Johansen, E.; Obis, D.; Pot, B.; Sanders, M.E.; Tremblay, A.; Ouwehand, A.C. Criteria to Qualify Microorganisms as “Probiotic” in Foods and Dietary Supplements. Front. Microbiol. 2020, 11, 1662. [Google Scholar] [CrossRef]
- Nogacka, A.; de los Reyes-Gavilán, C.G.; Arboleya, S.; Ruas-Madiedo, P.; Martínez-Faedo, C.; Suarez, A.; He, F.; Harata, G.; Endo, A.; Salazar, N.; et al. In Vitro Selection of Probiotics for Microbiota Modulation in Normal-Weight and Severely Obese Individuals: Focus on Gas Production and Interaction With Intestinal Epithelial Cells. Front. Microbiol. 2021, 12, 630572. [Google Scholar] [CrossRef] [PubMed]
- Swierz, M.J.; Storman, D.; Staskiewicz, W.; Gorecka, M.; Jasinska, K.W.; Swierz, A.M.; Tobola, P.; Skuza, A.; Bala, M.M. Efficacy of Probiotics in Patients with Morbid Obesity Undergoing Bariatric Surgery: A Systematic Review and Meta-Analysis. Surg. Obes. Relat. Dis. 2020, 16, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Aoun, A.; Darwish, F.; Hamod, N. The Influence of the Gut Microbiome on Obesity in Adults and the Role of Probiotics, Prebiotics, and Synbiotics for Weight Loss. Prev. Nutr. Food Sci. 2020, 25, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Nogacka, A.M.; Salazar, N.; Arboleya, S.; Ruas-Madiedo, P.; Mancabelli, L.; Suarez, A.; Martinez-Faedo, C.; Ventura, M.; Tochio, T.; Hirano, K.; et al. In Vitro Evaluation of Different Prebiotics on the Modulation of Gut Microbiota Composition and Function in Morbid Obese and Normal-Weight Subjects. Int. J. Mol. Sci. 2020, 21, 906. [Google Scholar] [CrossRef] [PubMed]
- Berglund, F.; Böhm, M.-E.; Martinsson, A.; Ebmeyer, S.; Österlund, T.; Johnning, A.; Larsson, D.G.J.; Kristiansson, E. Comprehensive Screening of Genomic and Metagenomic Data Reveals a Large Diversity of Tetracycline Resistance Genes. Microb. Genom. 2020, 6, mgen000455. [Google Scholar] [CrossRef]
- Montassier, E.; Valdés-Mas, R.; Batard, E.; Zmora, N.; Dori-Bachash, M.; Suez, J.; Elinav, E. Probiotics Impact the Antibiotic Resistance Gene Reservoir along the Human GI Tract in a Person-Specific and Antibiotic-Dependent Manner. Nat. Microbiol. 2021, 6, 1043–1054. [Google Scholar] [CrossRef]
- Lamberte, L.E.; van Schaik, W. Antibiotic Resistance in the Commensal Human Gut Microbiota. Curr. Opin. Microbiol. 2022, 68, 102150. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Gupta, S.S.; Nair, G.B.; Mande, S.S. In Silico Analysis of Antibiotic Resistance Genes in the Gut Microflora of Individuals from Diverse Geographies and Age-Groups. PLoS ONE 2013, 8, e83823. [Google Scholar] [CrossRef]
- Ahn, Y.; Jung, J.Y.; Kweon, O.; Veach, B.T.; Khare, S.; Gokulan, K.; Piñeiro, S.A.; Cerniglia, C.E. Impact of Chronic Tetracycline Exposure on Human Intestinal Microbiota in a Continuous Flow Bioreactor Model. Antibiotics 2021, 10, 886. [Google Scholar] [CrossRef]
- Relizani, K.; Le Corf, K.; Kropp, C.; Martin-Rosique, R.; Kissi, D.; Déjean, G.; Bruno, L.; Martinez, C.; Rawadi, G.; Elustondo, F.; et al. Selection of a Novel Strain of Christensenella Minuta as a Future Biotherapy for Crohn’s Disease. Sci. Rep. 2022, 12, 6017. [Google Scholar] [CrossRef]
- Mazier, W.; Le Corf, K.; Martinez, C.; Tudela, H.; Kissi, D.; Kropp, C.; Coubard, C.; Soto, M.; Elustondo, F.; Rawadi, G.; et al. A New Strain of Christensenella Minuta as a Potential Biotherapy for Obesity and Associated Metabolic Diseases. Cells 2021, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, M.A.H.M.; Palframan, R.J.; Cooper, J.M.; Gibson, G.R.; Rastall, R.A. In Vitro Fermentation of Sugar Beet Arabinan and Arabino-Oligosaccharides by the Human Gut Microflora. J. Appl. Microbiol. 2006, 100, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Jofre, J.; Muniesa, M. Antibiotic Resistance Genes in the Bacteriophage DNA Fraction of Environmental Samples. PLoS ONE 2011, 6, e17549. [Google Scholar] [CrossRef]
- Fouhy, F.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; Cotter, P.D. A Degenerate PCR-Based Strategy as a Means of Identifying Homologues of Aminoglycoside and β-Lactam Resistance Genes in the Gut Microbiota. BMC Microbiol. 2014, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.V.; Mellerup, A.; Christiansen, L.E.; Ståhl, M.; Olsen, J.E.; Angen, Ø. Sampling and Pooling Methods for Capturing Herd Level Antibiotic Resistance in Swine Feces Using QPCR and CFU Approaches. PLoS ONE 2015, 10, e0131672. [Google Scholar] [CrossRef]
- Quirós, P.; Colomer-Lluch, M.; Martínez-Castillo, A.; Miró, E.; Argente, M.; Jofre, J.; Navarro, F.; Muniesa, M. Antibiotic Resistance Genes in the Bacteriophage DNA Fraction of Human Fecal Samples. Antimicrob. Agents Chemother. 2014, 58, 606–609. [Google Scholar] [CrossRef]
- Gueimonde, M.; Salminen, S.; Isolauri, E. Presence of Specific Antibiotic (Tet) Resistance Genes in Infant Faecal Microbiota. FEMS Immunol. Med. Microbiol. 2006, 48, 21–25. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Level | Control | B. animalis IPLA20020 | B. bifidum TMC3108 | B. bifidum TMC3115 | B. longum IPLA20022 | L. rhamnosus GG |
|---|---|---|---|---|---|---|
| Bifidobacteriaceae | 4.47 ± 4.62 ab | 11.00 ± 2.92 b | 8.09 ± 5.65 ab | 8.03 ± 10.87 *ab | 2.08 ± 6.76 a | 1.75 ± 1.83 a |
| Coriobacteriaceae | 1.32 ± 1.04 * | 0.39 ± 1.25 | 0.89 ± 0.96 | −0.35 ± 1.38 | −0.04 ± 1.28 | −0.35 ± 2.16 |
| Bacteroidaceae | −0.89 ± 5.99 | −1.33 ± 2.71 * | −0.59 ± 1.54 | −1.17 ± 2.97 | 1.02 ± 3.83 | −2.75 ± 4.24 * |
| Rikenellaceae | 1.19 ± 2.19 | 0.33 ± 0.89 ** | 0.42 ± 0.73 | 0.20 ± 0.49 | 0.29 ± 0.57 * | 0.04 ± 0.33 |
| Tannerellaceae | 6.48 ± 3.67 | 3.33 ± 3.41 ** | 1.83 ± 3.40 | 1.50 ± 3.07 ** | 1.30 ± 2.97 * | 1.46 ± 3.51 * |
| Lactobacillaceae | 2.41 ± 6.08 a | 1.30 ± 3.93 a | 0.49 ± 0.17 ab | 2.99 ± 4.93 a | 2.05 ± 4.79 a | 19.34 ± 8.99 **b |
| Streptococcaceae | −0.16 ± 0.96 | −0.79 ± 1.14 | 0.18 ± 0.88 | −0.50 ± 1.00 | 0.11 ± 0.53 | −0.28 ± 0.28 |
| Christensenellaceae | −0.28 ± 0.26 * | −0.20 ± 0.33 | −0.09 ± 0.28 | −0.11 ± 0.09 | −0.08 ± 0.16 | −0.11 ± 0.19 |
| Clostridiaceae | −3.20 ± 3.47 | −2.40 ± 3.09 | −1.94 ± 2.94 | −2.69 ± 2.34 | −3.19 ± 3.70 | −3.10 ± 3.30 |
| Lachnospiraceae | −2.75 ± 6.46 | −3.76 ± 6.14 * | −2.86 ± 2.17 | −2.03 ± 7.08 | −2.40 ± 7.25 * | −3.94 ± 5.00 * |
| Ruminococcaceae | −1.82 ± 4.37 | −4.82 ± 5.67 ** | −4.61 ± 0.66 | −3.56 ± 4.53 | −1.29 ± 4.86 | −5.01 ± 5.81 * |
| Erysipelotrichaceae | 1.17 ± 4.83 * | 0.64 ± 3.18 | −0.92 ± 1.01 | 0.12 ± 1.12 | 0.37 ± 1.51 | −0.43 ± 1.85 |
| Acidaminococcaceae | 1.68 ± 3.74 | 3.32 ± 4.05 | 2.59 ± 4.56 | 3.74 ± 4.70 | 1.36 ± 1.49 | −1.04 ± 3.33 * |
| Veillonellaceae | −0.47 ± 1.96 | −0.28 ± 0.74 * | 1.27 ± 1.11 | 0.06 ± 0.92 * | −0.13 ± 0.34 | −0.29 ± 1.05 |
| Burkholderiaceae | −0.84 ± 0.94 | −1.12 ± 1.15 | −0.46 ± 0.58 | −0.82 ± 1.20 | −0.67 ± 0.86 | −1.04 ± 1.09 |
| Enterobacteriaceae | −6.28 ± 3.73 * | −3.68 ± 1.90 ** | −3.55 ± 3.85 | −3.21 ± 4.71 ** | −1.54 ± 3.55 * | −4.37 ± 3.66 ** |
| Family Level | Control | B. animalis IPLA20020 | B. bifidum TMC3108 | B. bifidum TMC3115 | B. longum IPLA20022 | L. rhamnosus GG |
|---|---|---|---|---|---|---|
| Bifidobacteriaceae | 1.66 ± 2.54 * | 4.19 ± 8.01 * | 6.55 ± 4.62 | 4.26 ± 4.90 | 0.31 ± 5.90 | 1.32 ± 2.48 * |
| Coriobacteriaceae | 3.30 ± 4.17 | 0.78 ± 1.14 | 0.18 ± 0.31 | 0.61 ± 1.70 | 0.62 ± 1.48 | 2.37 ± 2.65 |
| Bacteroidaceae | −2.02 ± 6.83 | −1.66 ± 1.74 | −1.59 ± 1.80 | 0.74 ± 3.93 | 2.86 ± 5.43 | −2.78 ± 3.19 |
| Rikenellaceae | 0.62 ± 0.89 | 0.84 ± 1.31 | 0.06 ± 0.72 | 0.22 ± 0.56 | 0.54 ± 0.62 | 0.11 ± 0.70 |
| Tannerellaceae | 3.99 ± 4.57 * | 4.95 ± 2.88 * | 2.35 ± 0.30 | 3.07 ± 2.43 | 5.41 ± 6.92 | 2.09 ± 2.21 |
| Lactobacillaceae | 0.04 ± 0.12 a | 0.08 ± 0.18 a | 0.01 ± 0.01 ab | 0.05 ± 0.15 *a | 0.19 ± 0.31 a | 12.89 ± 5.86 *b |
| Streptococcaceae | −1.55 ± 6.12 | −0.25 ± 2.68 | 0.55 ± 0.72 | −0.39 ± 1.95 | 0.30 ± 0.88 | −0.53 ± 1.64 * |
| Christensenellaceae | −1.71 ± 1.88 * | −0.12 ± 1.15 | −0.14 ± 0.88 | −0.65 ± 0.94 * | 0.14 ± 1.54 | −1.01 ± 1.74 |
| Clostridiaceae | 0.39 ± 1.04 * | 0.90 ± 3.17 | −0.15 ± 0.15 | 0.79 ± 2.63 * | 1.52 ± 4.76 | −0.31 ± 0.60 * |
| Lachnospiraceae | −0.92 ± 4.62 | −2.91 ± 2.72 | −1.15 ± 3.41 | −2.12 ± 3.28 | −2.04 ± 3.69 | −3.24 ± 2.75 * |
| Ruminococcaceae | 2.27 ± 10.63 | −4.02 ± 3.85 | −3.08 ± 3.51 | −2.12 ± 3.92 | −0.98 ± 6.77 | −5.06 ± 4.35 |
| Erysipelotrichaceae | 1.31 ± 1.74 | 2.03 ± 2.70 | 0.78 ± 1.95 | 1.10 ± 1.78 | 1.38 ± 2.33 | 0.73 ± 1.68 |
| Acidaminococcaceae | −1.17 ± 2.08 | −0.42 ± 1.59 | −0.58 ± 0.99 | −0.52 ± 1.02 | −0.54 ± 1.38 * | −1.65 ± 1.89 |
| Veillonellaceae | 0.35 ± 0.77 | 0.34 ± 0.68 | 0.14 ± 0.17 | 0.37 ± 0.65 | 0.43 ± 0.63 | 0.36 ± 0.98 |
| Burkholderiaceae | 0.91 ± 1.76 | 0.49 ± 1.04 * | −0.14 ± 0.32 | 0.12 ± 0.74 | 0.31 ± 0.62 * | −0.14 ± 0.89 * |
| Enterobacteriaceae | −5.78 ± 7.55 * | −4.49 ± 3.00 * | −2.25 ± 2.89 | −4.40 ± 3.12 | 0.33 ± 9.71 | −4.13 ± 3.59 * |
| ARG | Gene | Primer | Sequence 5′–3′ | Size (pb) | Tª (°C) | Ref |
|---|---|---|---|---|---|---|
| β-lactams | blaSHV | bla-SHV F PCR | CACTCAAGGATGTATTGTG | 885 | 58 | [47] |
| bla-SHV R PCR | TTAGCGTTGCCAGTGCTCG | |||||
| bla-SHV F qPCR | GCTGGAGCGAAAGATCCACT | 247 | 60 | [48] | ||
| bla-SHV R qPCR | CGCCTCATTCAGTTCCGTTT | |||||
| blaTEM | bla-TEM F | CACTATTCTCAGAATGACTTGGT | 85 | 60 | [49] | |
| bla-TEM R | TGCATAATTCTCTTACTGTCATG | |||||
| Tetracyclines | tetO | tetO F | ACGGARAGTTTATTGTATACC | 171 | 60 | [50] |
| tetO R | TGGCGTATCTATAATGTTGAC | |||||
| tetM | tetM F | ACAGAAAGCTTATTATATAAC | 171 | 55 | [50] | |
| tetM R | TGGCGTGTCTATGATGTTCAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogacka, A.M.; Saturio, S.; Alvarado-Jasso, G.M.; Salazar, N.; de los Reyes Gavilán, C.G.; Martínez-Faedo, C.; Suarez, A.; Wang, R.; Miyazawa, K.; Harata, G.; et al. Probiotic-Induced Modulation of Microbiota Composition and Antibiotic Resistance Genes Load, an In Vitro Assessment. Int. J. Mol. Sci. 2024, 25, 1003. https://doi.org/10.3390/ijms25021003
Nogacka AM, Saturio S, Alvarado-Jasso GM, Salazar N, de los Reyes Gavilán CG, Martínez-Faedo C, Suarez A, Wang R, Miyazawa K, Harata G, et al. Probiotic-Induced Modulation of Microbiota Composition and Antibiotic Resistance Genes Load, an In Vitro Assessment. International Journal of Molecular Sciences. 2024; 25(2):1003. https://doi.org/10.3390/ijms25021003
Chicago/Turabian StyleNogacka, Alicja Maria, Silvia Saturio, Guadalupe Monserrat Alvarado-Jasso, Nuria Salazar, Clara G. de los Reyes Gavilán, Ceferino Martínez-Faedo, Adolfo Suarez, Ruipeng Wang, Kenji Miyazawa, Gaku Harata, and et al. 2024. "Probiotic-Induced Modulation of Microbiota Composition and Antibiotic Resistance Genes Load, an In Vitro Assessment" International Journal of Molecular Sciences 25, no. 2: 1003. https://doi.org/10.3390/ijms25021003
APA StyleNogacka, A. M., Saturio, S., Alvarado-Jasso, G. M., Salazar, N., de los Reyes Gavilán, C. G., Martínez-Faedo, C., Suarez, A., Wang, R., Miyazawa, K., Harata, G., Endo, A., Arboleya, S., & Gueimonde, M. (2024). Probiotic-Induced Modulation of Microbiota Composition and Antibiotic Resistance Genes Load, an In Vitro Assessment. International Journal of Molecular Sciences, 25(2), 1003. https://doi.org/10.3390/ijms25021003