
Expanded Gene Regulatory Network Reveals Potential Light-Responsive Transcription Factors and Target Genes in Cordyceps militaris


 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
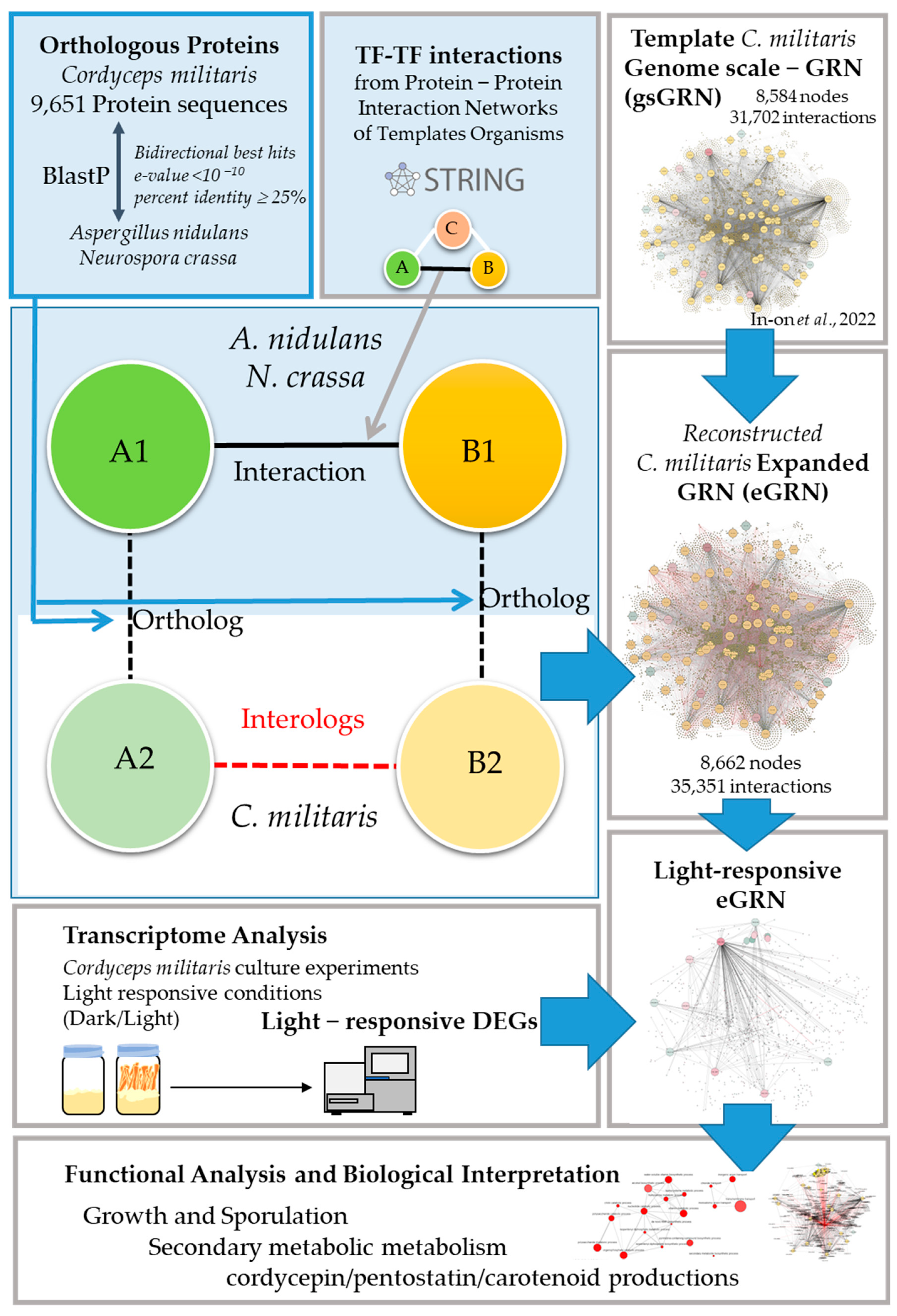
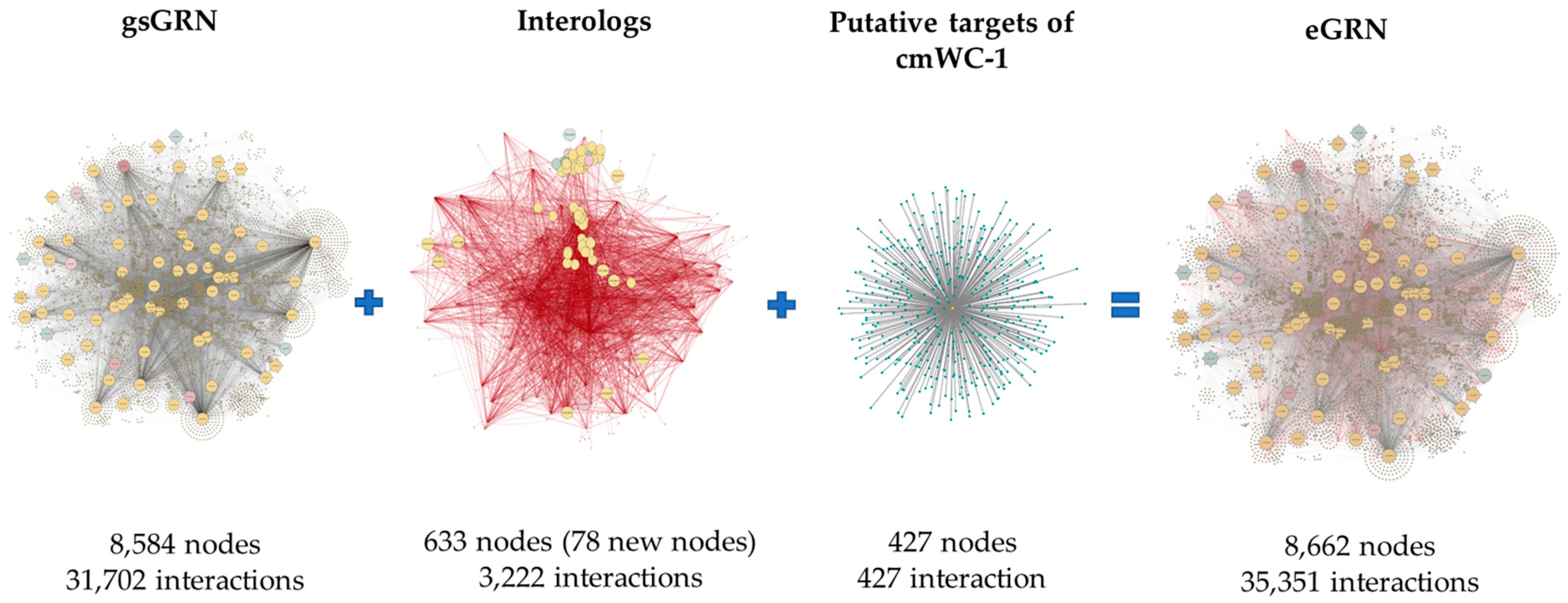
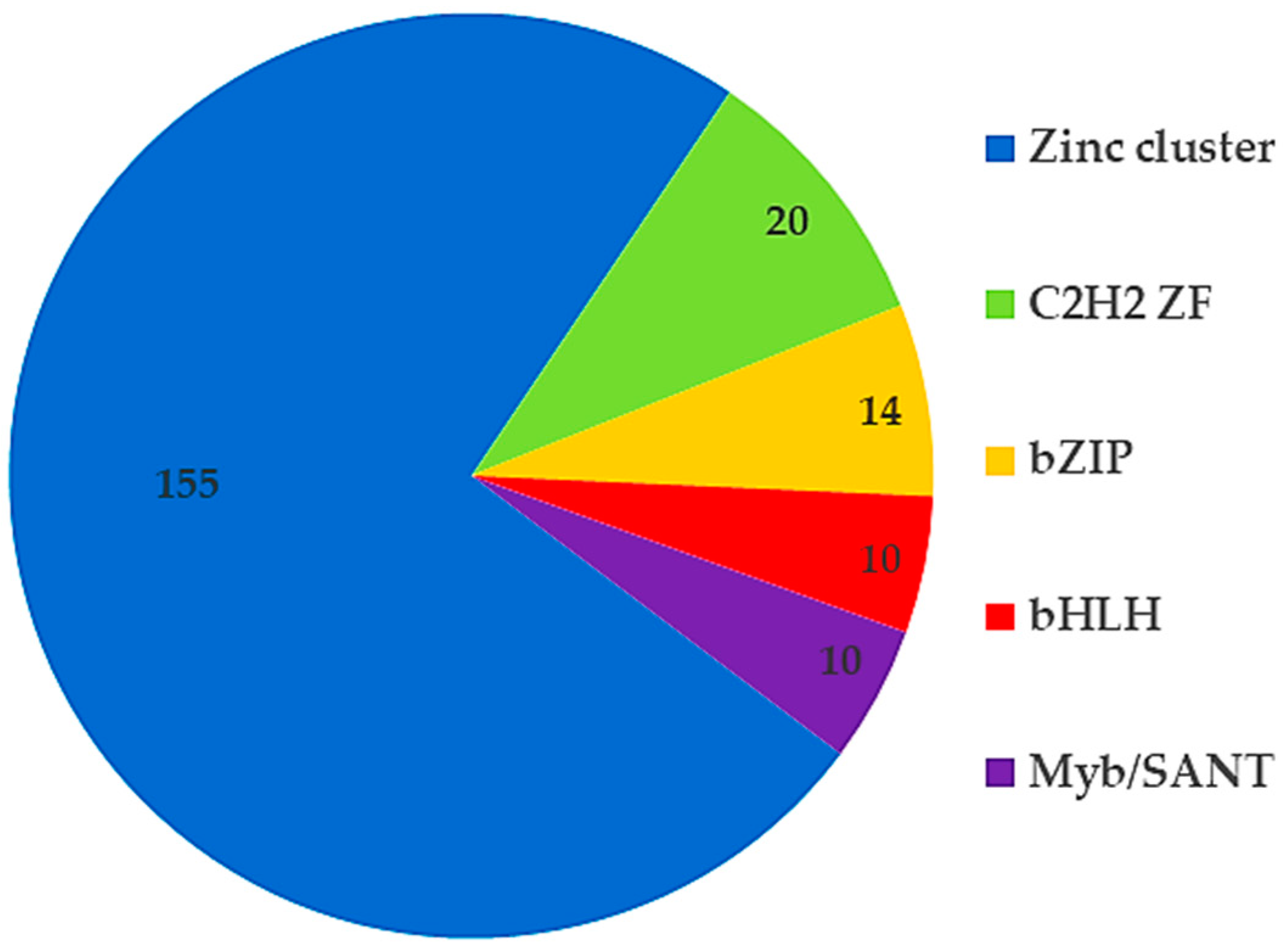
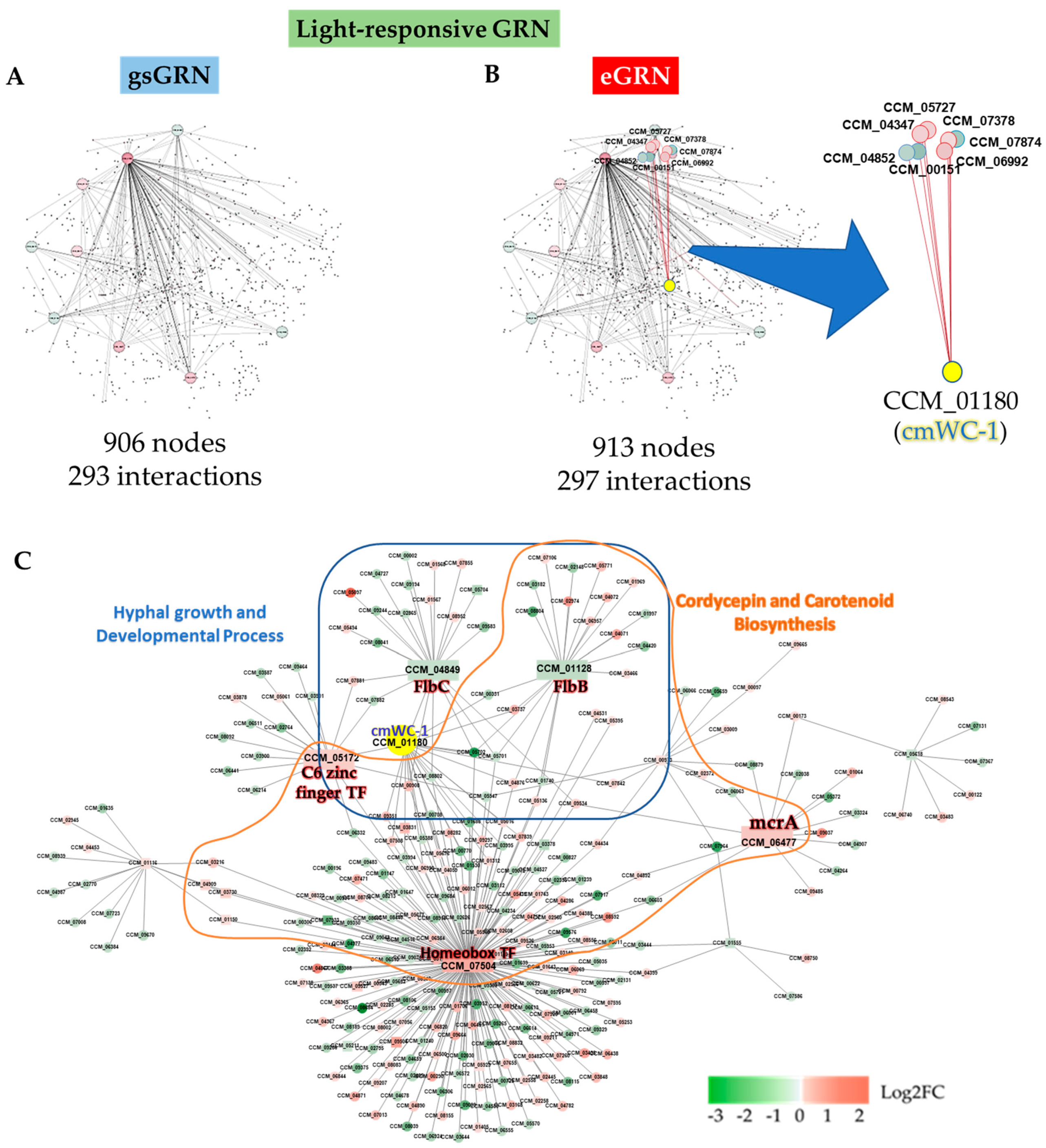
2.1. The Reconstructed eGRN of C. militaris
2.2. Growth Characteristics and Production of Cordycepin and Carotenoids
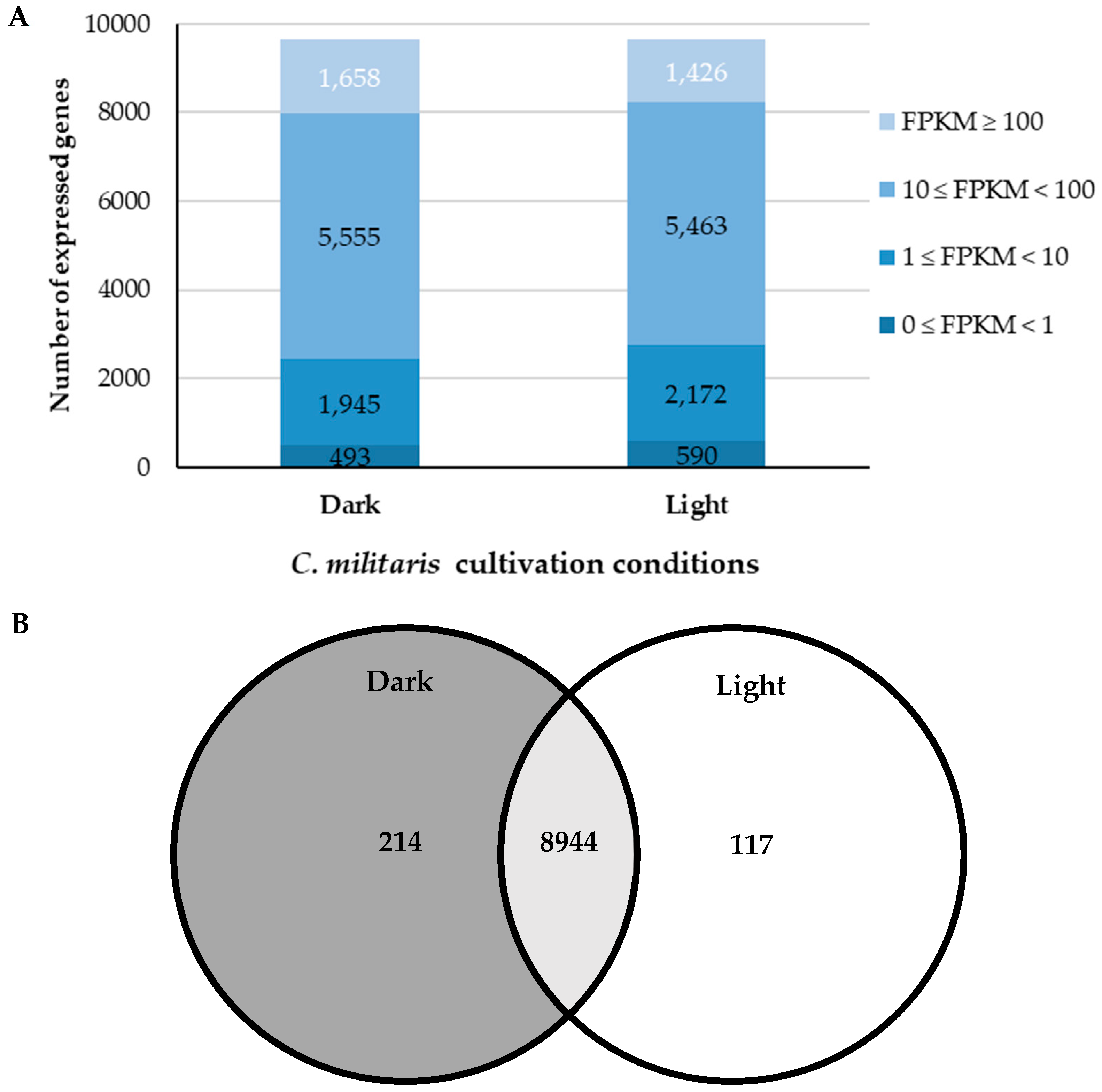
2.3. Comparative Transcriptome Analysis of C. militaris under Dark and Light Conditions
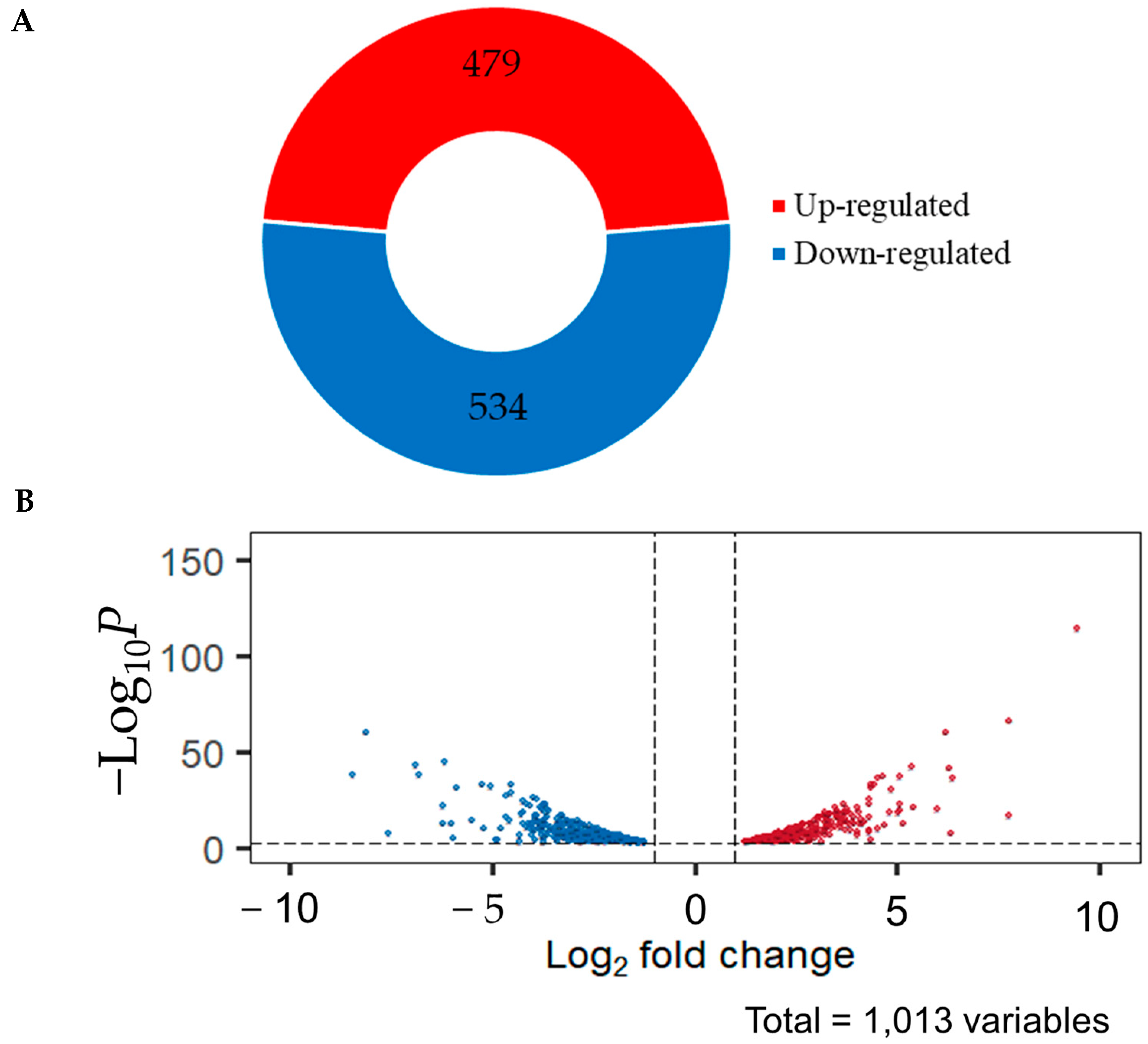
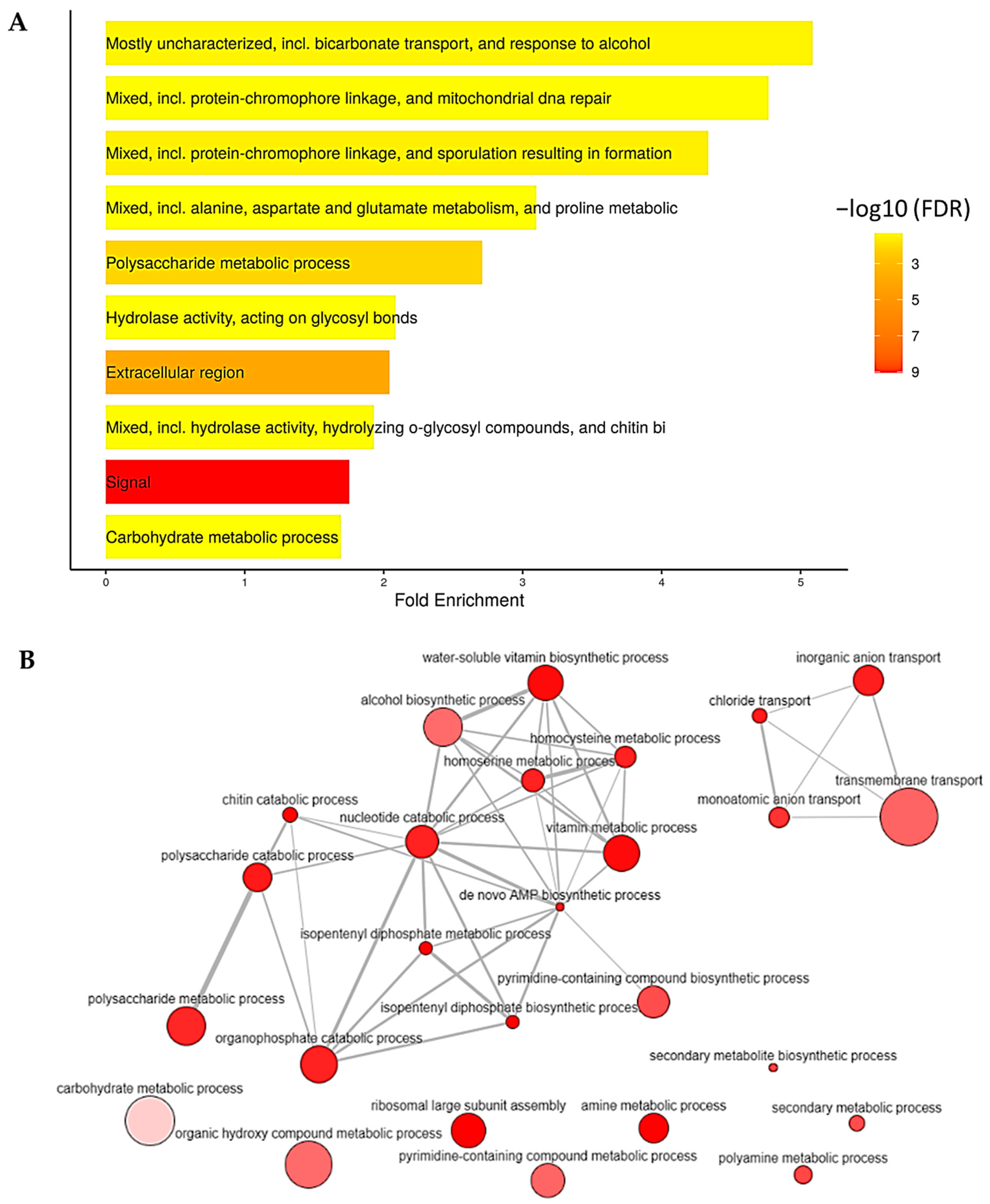
2.4. Identified Differentially Expressed Genes (DEGs)
2.5. Identified Light-Responsive Genes and Potential TFs Using the Reconstructed eGRN
2.6. Light-Responsive Genes and Potential TFs Involving Hyphal Growth, Sporulation, Biosynthesis of Cordycepin and Carotenoids in C. militaris
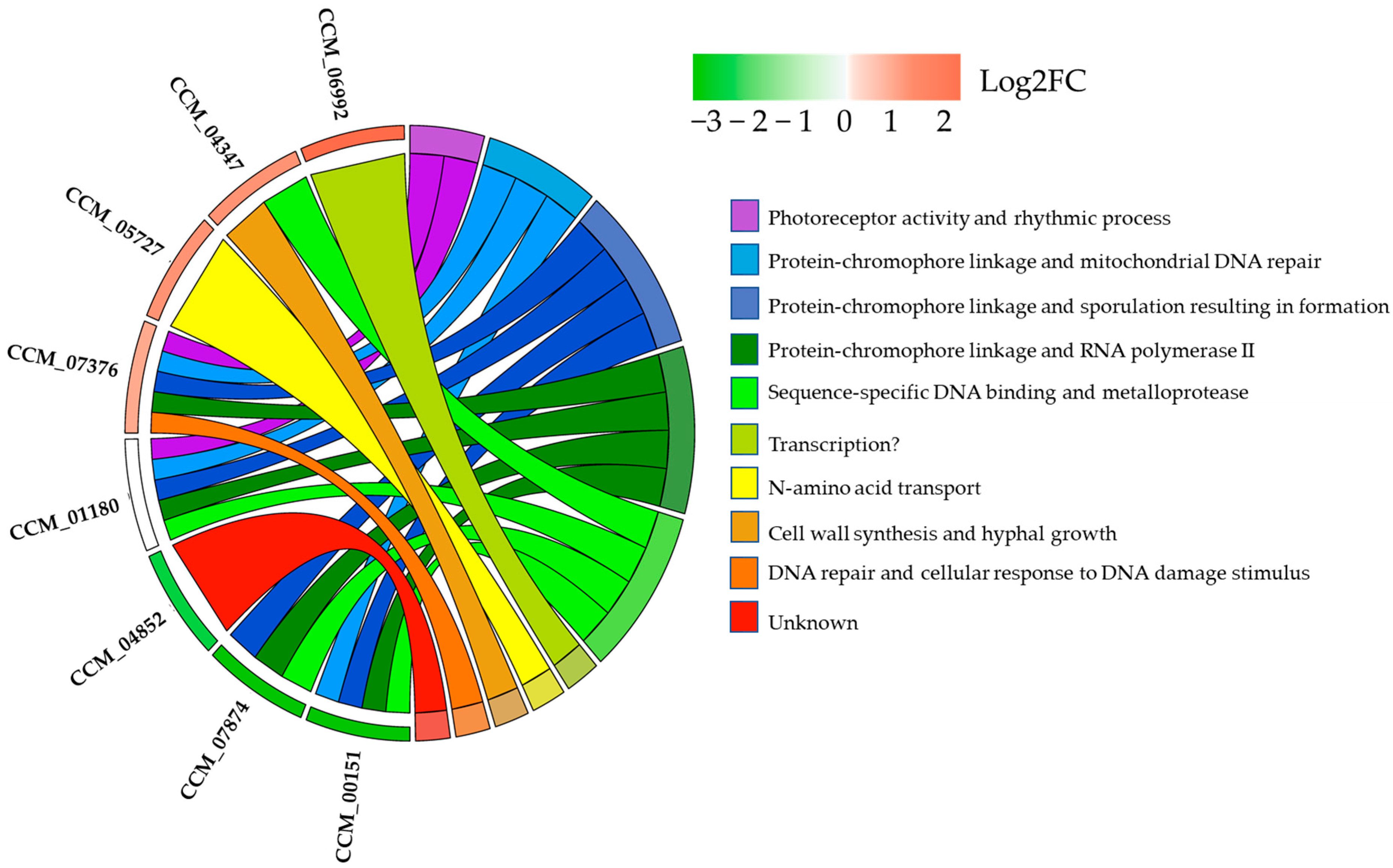
2.7. The Potential Physical Interaction of the CmWC-1 and Its Candidate Protein Targets
3. Materials and Methods
3.1. Expanding Gene Regulatory Network (eGRN) of C. militaris
3.2. Transcriptome Analysis of C. militaris
3.3. Identification of Light-Responsive Genes and Potential TFs Using the eGRN of C. militaris
3.4. Molecular Docking Simulation of the CmWC-1 with Its Potential Protein Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, T.; Li, G.; Kang, J.; Kang, C.; Hyde, K.D. Optimization of solid-state fermentation for fruiting body growth and cordycepin production by Cordyceps militaris. Chiang Mai J. Sci. 2014, 41, 858–872. [Google Scholar]
- Das, S.K.; Masuda, M.; Sakurai, A.; Sakakibara, M. Medicinal uses of the mushroom Cordyceps militaris: Current state and prospects. Fitoterapia 2010, 81, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Zhang, W.; Zhang, Y.; Liu, X. The medicinal fungus Cordyceps militaris: Research and development. Mycol. Prog. 2012, 11, 599–614. [Google Scholar] [CrossRef]
- Adnan, M.; Ashraf, S.A.; Khan, S.; Alshammari, E.; Awadelkareem, A.M. Effect of pH, temperature and incubation time on cordycepin production from Cordyceps militaris using solid-state fermentation on various substrates. CyTA-J. Food 2017, 15, 617–621. [Google Scholar] [CrossRef]
- Shih, L.; Tsai, K.-L.; Hsieh, C. Effects of culture conditions on the mycelial growth and bioactive metabolite production in submerged culture of Cordyceps militaris. Biochem. Eng. J. 2007, 33, 193–201. [Google Scholar] [CrossRef]
- Chiang, S.-S.; Liang, Z.-C.; Wang, Y.-C.; Liang, C.-H. Effect of light-emitting diodes on the production of cordycepin, mannitol and adenosine in solid-state fermented rice by Cordyceps militaris. J. Food Compos. Anal. 2017, 60, 51–56. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Luo, F.; Wang, C. Bioactive metabolites and potential mycotoxins produced by Cordyceps fungi: A review of safety. Toxins 2020, 12, 410. [Google Scholar] [CrossRef]
- Yu, Z.; Fischer, R. Light sensing and responses in fungi. Nat. Rev. Microbiol. 2019, 17, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, F.; Lan, N.; Liu, B.; Hu, C.; Xue, W.; Zhang, Z.; Li, S. The Zn (II) 2Cys6-type transcription factor ADA-6 regulates conidiation, sexual development, and oxidative stress response in Neurospora crassa. Front. Microbiol. 2019, 10, 750. [Google Scholar] [CrossRef]
- Yang, T.; Guo, M.; Yang, H.; Guo, S.; Dong, C. The blue-light receptor CmWC-1 mediates fruit body development and secondary metabolism in Cordyceps militaris. Appl. Microbiol. Biotechnol. 2016, 100, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2012, 12, R116. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yu, G.; Chen, Y.; Jiang, S.; Wang, M.; Jin, Y.; Lan, X.; Liang, Y.; Sun, H. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. PLoS ONE 2012, 7, e51853. [Google Scholar] [CrossRef] [PubMed]
- Thananusak, R.; Laoteng, K.; Raethong, N.; Zhang, Y.; Vongsangnak, W. Metabolic responses of carotenoid and cordycepin biosynthetic pathways in Cordyceps militaris under light-programming exposure through genome-wide transcriptional analysis. Biology 2020, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- In-On, A.; Thananusak, R.; Ruengjitchatchawalya, M.; Vongsangnak, W.; Laomettachit, T. Construction of light-responsive gene regulatory network for growth, development and secondary metabolite production in Cordyceps militaris. Biology 2022, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, F.; Liu, M.; Fu, M.; Dong, C. Dynamic genome-wide transcription profiling and direct target genes of CmWC-1 reveal hierarchical light signal transduction in Cordyceps militaris. J. Fungi 2022, 8, 624. [Google Scholar] [CrossRef]
- MacPherson, S.; Larochelle, M.; Turcotte, B. A fungal family of transcriptional regulators: The zinc cluster proteins. Microbiol. Mol. Biol. Rev. MMBR 2006, 70, 583–604. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Shlomi, T.; Feizi, H.; Ideker, T.; Sharan, R. Transcriptional regulation of protein complexes within and across species. Proc. Natl. Acad. Sci. USA 2007, 104, 1283–1288. [Google Scholar] [CrossRef]
- Feng, Y.-j.; Zhu, Y.; Li, Y.-m.; Li, J.; Sun, Y.-f.; Shen, H.-t.; Wang, A.-y.; Lin, Z.-p.; Zhu, J.-b. Effect of strain separated parts, solid-state substrates and light condition on yield and bioactive compounds of Cordyceps militaris fruiting bodies. CyTA-J. Food 2018, 16, 916–922. [Google Scholar] [CrossRef]
- Dong, J.Z.; Lei, C.; Zheng, X.J.; Ai, X.R.; Wang, Y.; Wang, Q. Light wavelengths regulate growth and active components of Cordyceps militaris fruit bodies. J. Food Biochem. 2013, 37, 578–584. [Google Scholar] [CrossRef]
- Suparmin, A.; Kato, T.; Takemoto, H.; Park, E.Y. Metabolic comparison of aerial and submerged mycelia formed in the liquid surface culture of Cordyceps militaris. MicrobiologyOpen 2019, 8, e00836. [Google Scholar] [CrossRef]
- Son, S.-H.; Son, Y.-E.; Cho, H.-J.; Chen, W.; Lee, M.-K.; Kim, L.-H.; Han, D.-M.; Park, H.-S. Homeobox proteins are essential for fungal differentiation and secondary metabolism in Aspergillus nidulans. Sci. Rep. 2020, 10, 6094. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.S.; Zheng, J.; Yin, Y.; Lorber, S.; Puel, O.; Dhingra, S.; Espeso, E.A.; Calvo, A.M. Homeobox transcription factor HbxA influences expression of over one thousand genes in the model fungus Aspergillus nidulans. PLoS ONE 2023, 18, e0286271. [Google Scholar] [CrossRef]
- Son, S.-H.; Lee, M.-K.; Son, Y.-E.; Park, H.-S. HbxB is a key regulator for stress response and β-glucan biogenesis in Aspergillus nidulans. Microorganisms 2021, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Dabholkar, A.; Wyman, E.; Lohmar, J.; Cary, J. Regulatory functions of homeobox domain transcription factors in fungi. Appl. Environ. Microbiol. 2024, 90, e02208–e02223. [Google Scholar] [CrossRef]
- Kwon, N.J.; Garzia, A.; Espeso, E.A.; Ugalde, U.; Yu, J.H. FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol. Microbiol. 2010, 77, 1203–1219. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Q.; Zhang, J.; Liu, K.; Li, K.; Liu, G.; Dong, C. Comparative transcriptome analysis between a spontaneous albino mutant and its sibling strain of Cordyceps militaris in response to light stress. Front. Microbiol. 2018, 9, 1237. [Google Scholar] [CrossRef]
- Otamendi, A.; Perez-de-Nanclares-Arregi, E.; Oiartzabal-Arano, E.; Cortese, M.S.; Espeso, E.A.; Etxebeste, O. Developmental regulators FlbE/D orchestrate the polarity site-to-nucleus dynamics of the fungal bZIP transcription factor FlbB. Cell. Mol. Life Sci. CMLS 2019, 76, 4369–4390. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.K.; Ehrlich, K.C. Genome-wide analysis of the Zn(II)(2)Cys(6) zinc cluster-encoding gene family in Aspergillus flavus. Appl. Microbiol. Biotechnol. 2013, 97, 4289–4300. [Google Scholar] [CrossRef]
- Muñoz-Guzmán, F.; Caballero, V.; Larrondo, L.F. A global search for novel transcription factors impacting the Neurospora crassa circadian clock. G3 Genes|Genomes|Genet. 2021, 11, jkab100. [Google Scholar] [CrossRef]
- Oakley, C.E.; Ahuja, M.; Sun, W.W.; Entwistle, R.; Akashi, T.; Yaegashi, J.; Guo, C.J.; Cerqueira, G.C.; Russo Wortman, J.; Wang, C.C.; et al. Discovery of McrA, a master regulator of Aspergillus secondary metabolism. Mol. Microbiol. 2017, 103, 347–365. [Google Scholar] [CrossRef]
- Kunhorm, P.; Chaicharoenaudomrung, N.; Noisa, P. Enrichment of cordycepin for cosmeceutical applications: Culture systems and strategies. Appl. Microbiol. Biotechnol. 2019, 103, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.-W.; Zhao, Y.; Tang, H.-B.; Ye, Z.-W.; Wei, T.; Lin, J.-F.; Guo, L.-Q. Transcriptome analysis of Cordyceps militaris reveals genes associated with carotenoid synthesis and identification of the function of the cmtns gene. Front. Microbiol. 2019, 10, 2105. [Google Scholar] [CrossRef]
- Patil, A.; Kumagai, Y.; Liang, K.-c.; Suzuki, Y.; Nakai, K. Linking transcriptional changes over time in stimulated dendritic cells to identify gene networks activated during the innate immune response. PLoS Comput. Biol. 2013, 9, e1003323. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Park, J.; Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Kim, S.; et al. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef]
- Thananusak, R.; Laoteng, K.; Raethong, N.; Koffas, M.; Vongsangnak, W. Dissecting metabolic regulation in mycelial growth and fruiting body developmental stages of Cordyceps militaris through integrative transcriptome analysis. Biotechnol. Bioprocess Eng. 2022, 28, 406–418. [Google Scholar] [CrossRef]
- Yang, T.; Sun, J.; Lian, T.; Wang, W.; Dong, C. Process Optimization for Extraction of Carotenoids from Medicinal Caterpillar Fungus, Cordyceps militaris (Ascomycetes). Int. J. Med. Mushrooms 2014, 16, 125–135. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 31 January 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Biomass (g kg−1) | Cordycepin Content (mg g−1 Cell Mass) | Carotenoid Content (mg g−1 Cell Mass) |
|---|---|---|---|
| Dark | 57.857 ± 4.67 | 13.014 ± 0.04 | 0.092 ± 0.05 |
| Light | 74.376 ± 3.35 * | 23.294 ± 0.80 * | 1.916 ± 0.09 * |
| Sample | Total Clean Reads (M) | GC (%) | Uniquely Mapped Reads (M) | Percentage of Uniquely Mapped Reads |
|---|---|---|---|---|
| D-1 | 24.88 | 56.00 | 23.81(95.71%) | 95.71 |
| D-2 | 19.96 | 56.00 | 19.05 (95.48%) | 95.46 |
| L-1 | 22.25 | 56.00 | 21.32 (95.84%) | 95.84 |
| L-2 | 24.48 | 57.00 | 23.50 (95.99%) | 95.99 |
| Average | 22.89 ± 1.76 | 56.25 ± 0.39 | 21.92 ± 1.71 | 95.76 ± 0.19 |
| Gene ID | TFs | DEGs | Number of Regulated Genes by TFs | Normalized Betweenness Score |
|---|---|---|---|---|
| CCM_07504 | Homeobox transcription factor | Up | 186 | 1.00 |
| CCM_04849 | C2H2 ZF transcription factor (FlbC) | Down | 21 | 0.11 |
| CCM_01128 | bZIP-type transcription factor (FlbB) | Down | 20 | 0.12 |
| CCM_05172 | C6 zinc finger domain containing protein | Up | 16 | 0.10 |
| CCM_06477 | C6 transcription factor (mcrA) | Up | 14 | 0.13 |
| Ligands (Target Proteins) | CCM_ 00151 | CCM_ 07874 | CCM_ 04852 | CCM_ 07378 | CCM_ 05727 | CCM_ 04347 | CCM_ 06992 | CCM_ 00072 | CCM_ 01336 |
|---|---|---|---|---|---|---|---|---|---|
| Docking Score | −273.95 | −222.17 | −242.87 | −239.55 | −330.6 | −254.37 | −289.5 | −301.36 | −262.15 |
| Confidence Score | 0.923 | 0.809 | 0.865 | 0.857 | 0.974 | 0.890 | 0.942 | 0.954 | 0.904 |
| Ligand RMSD (Å) | 65.63 | 56.07 | 45.19 | 44.50 | 59.91 | 59.36 | 38.27 | 79.80 | 79.58 |
| Template receptor (PDB ID) | 4F3L | 4F3L | 4F3L | 4F3L | 4F3L | 4F3L | 4F3L | 4F3L | 4F3L |
| Template ligand (PDB ID) | 1DNP | 2L2F | 5A8I | 3P8A | 3GIA | 5D3Q | - | 4M4X | 2LDU |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buradam, P.; Thananusak, R.; Koffas, M.; Chumnanpuen, P.; Vongsangnak, W. Expanded Gene Regulatory Network Reveals Potential Light-Responsive Transcription Factors and Target Genes in Cordyceps militaris. Int. J. Mol. Sci. 2024, 25, 10516. https://doi.org/10.3390/ijms251910516
Buradam P, Thananusak R, Koffas M, Chumnanpuen P, Vongsangnak W. Expanded Gene Regulatory Network Reveals Potential Light-Responsive Transcription Factors and Target Genes in Cordyceps militaris. International Journal of Molecular Sciences. 2024; 25(19):10516. https://doi.org/10.3390/ijms251910516
Chicago/Turabian StyleBuradam, Paradee, Roypim Thananusak, Mattheos Koffas, Pramote Chumnanpuen, and Wanwipa Vongsangnak. 2024. "Expanded Gene Regulatory Network Reveals Potential Light-Responsive Transcription Factors and Target Genes in Cordyceps militaris" International Journal of Molecular Sciences 25, no. 19: 10516. https://doi.org/10.3390/ijms251910516
APA StyleBuradam, P., Thananusak, R., Koffas, M., Chumnanpuen, P., & Vongsangnak, W. (2024). Expanded Gene Regulatory Network Reveals Potential Light-Responsive Transcription Factors and Target Genes in Cordyceps militaris. International Journal of Molecular Sciences, 25(19), 10516. https://doi.org/10.3390/ijms251910516