
Molecular Long-Term Analysis of the GMMG-HD4 Trial in Multiple Myeloma—Patterns of Association of Chromosomal Aberrations with Response and Proliferation Determining Survival in Selecting Treatments in View of Limited Resources in Low- and Middle-Income Countries

 , , , , , , ,
, , , , , , ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Prognostic Factors for Long-Term Survival—Response, Proliferation, and Molecular Alterations
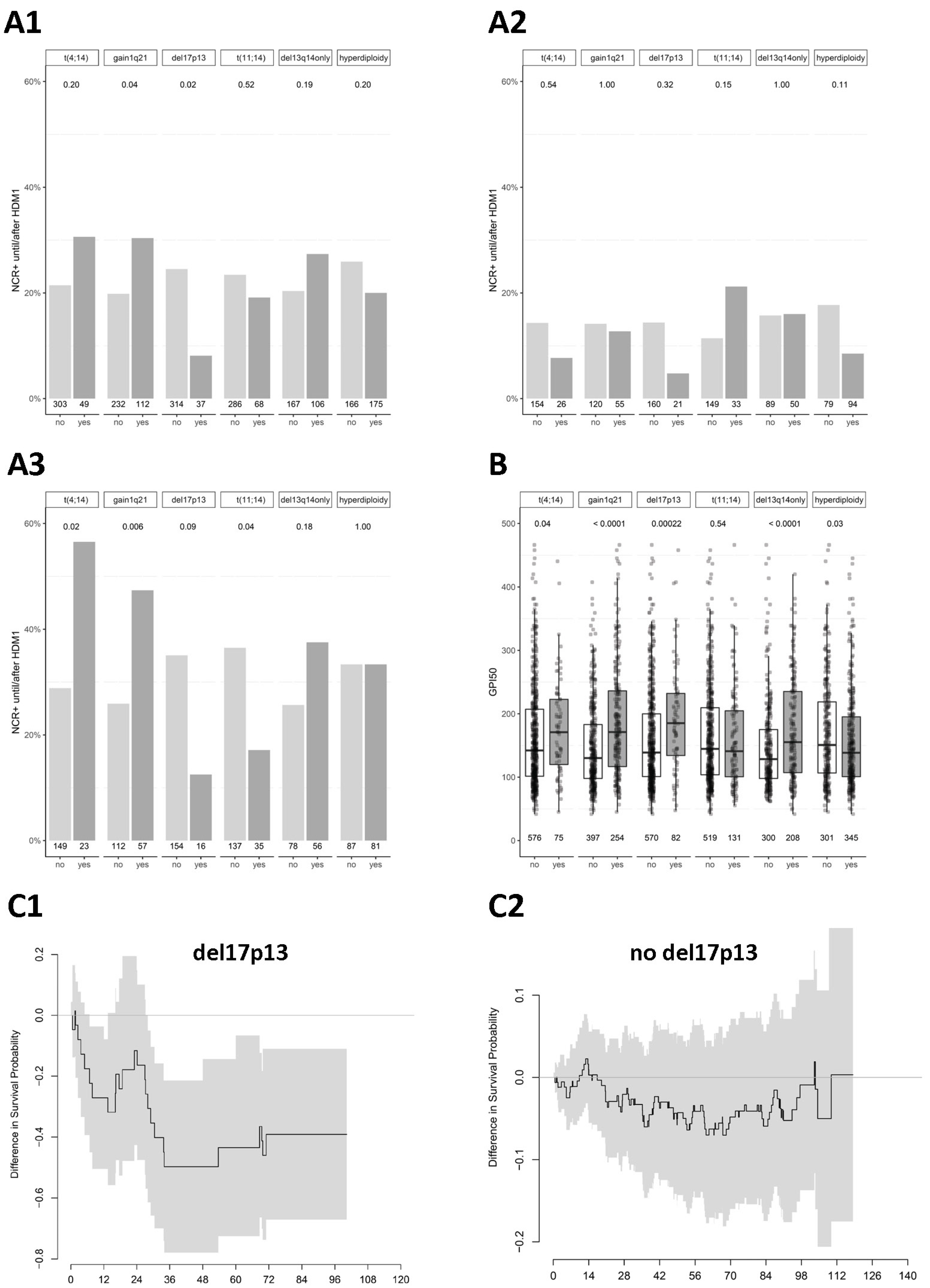
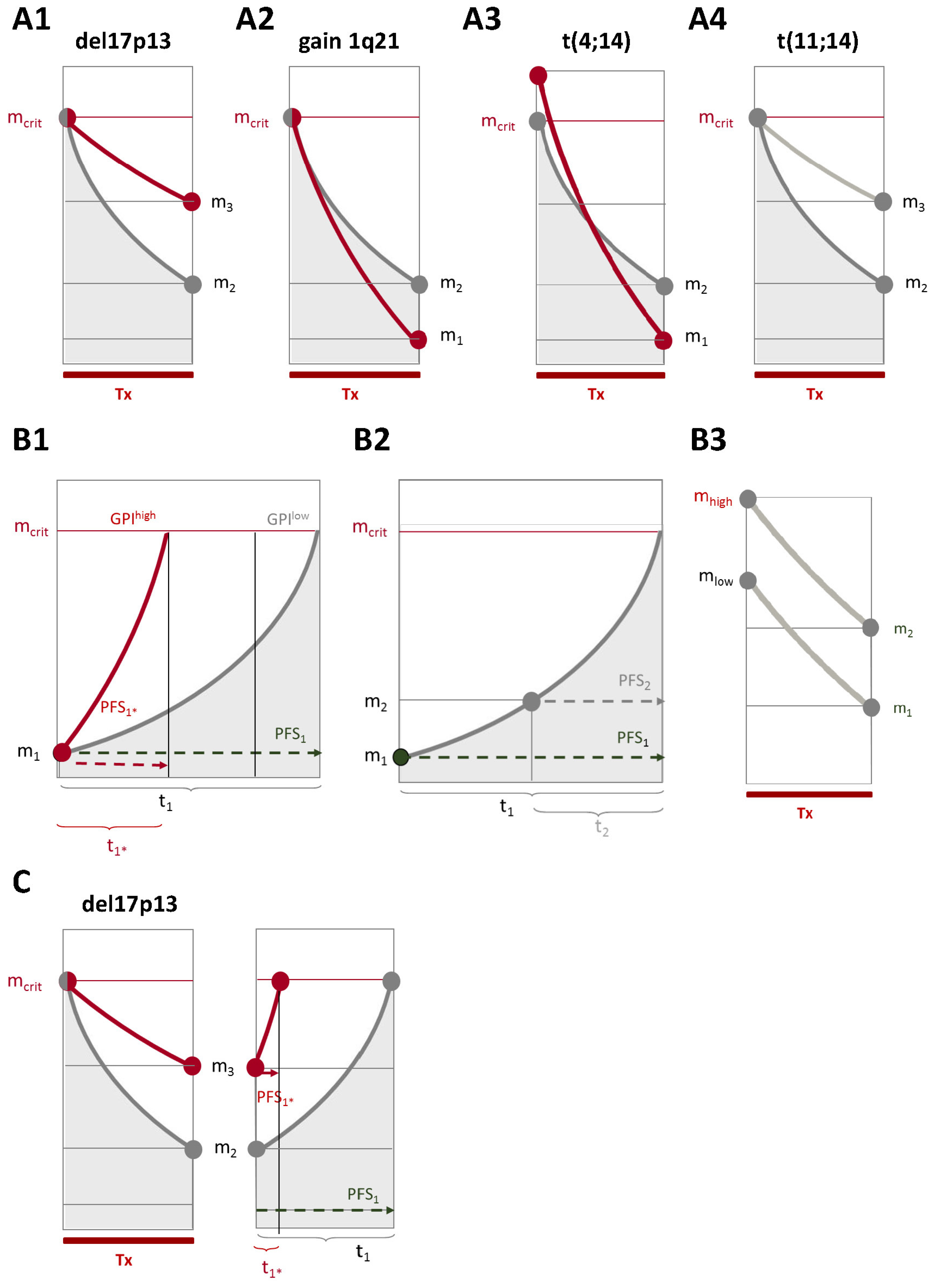
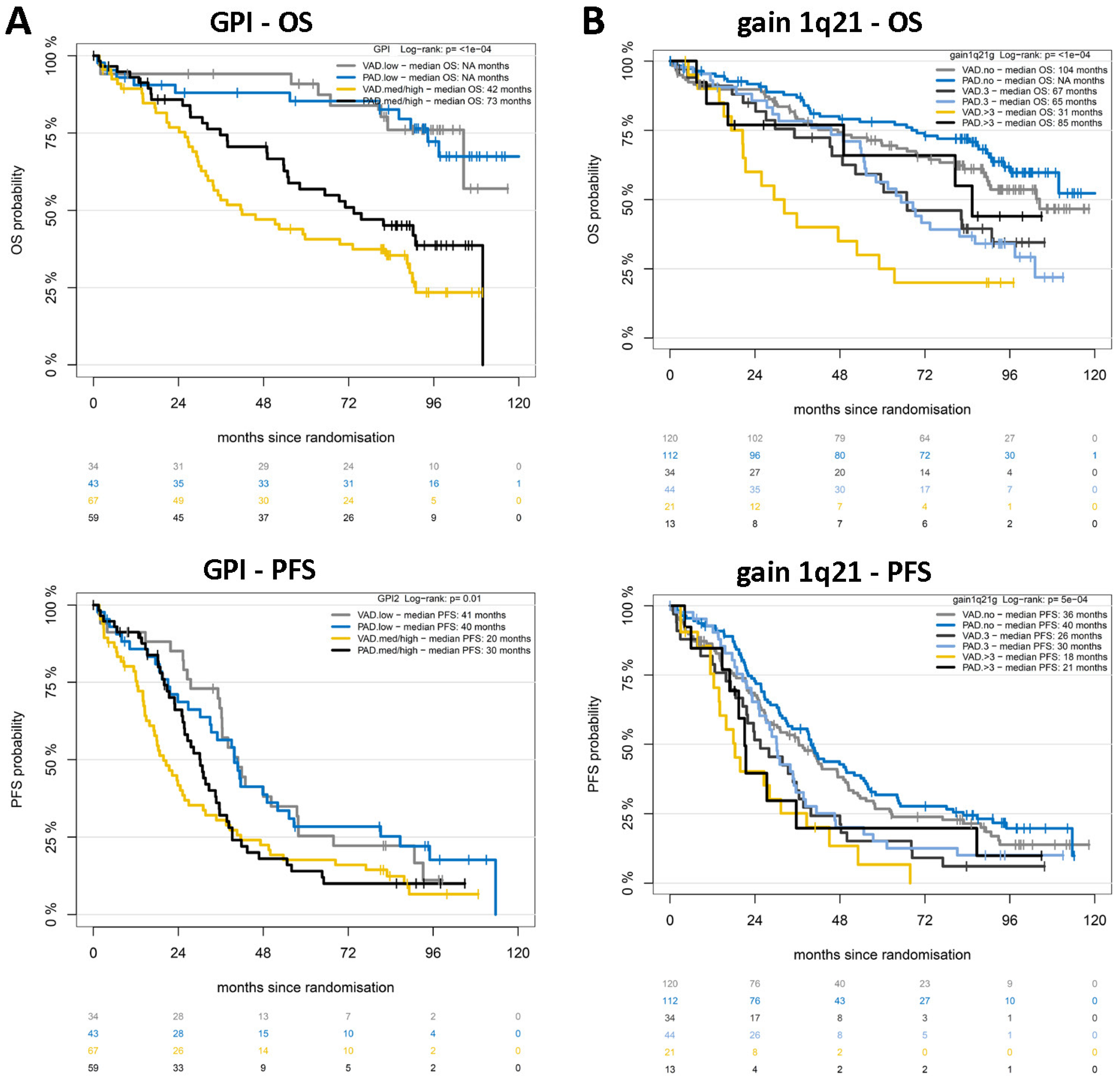
2.2. Chromosomal Aberrations Determine Long-Term Prognosis by Different Patterns of Association with Proliferation and Treatment-Dependent Response
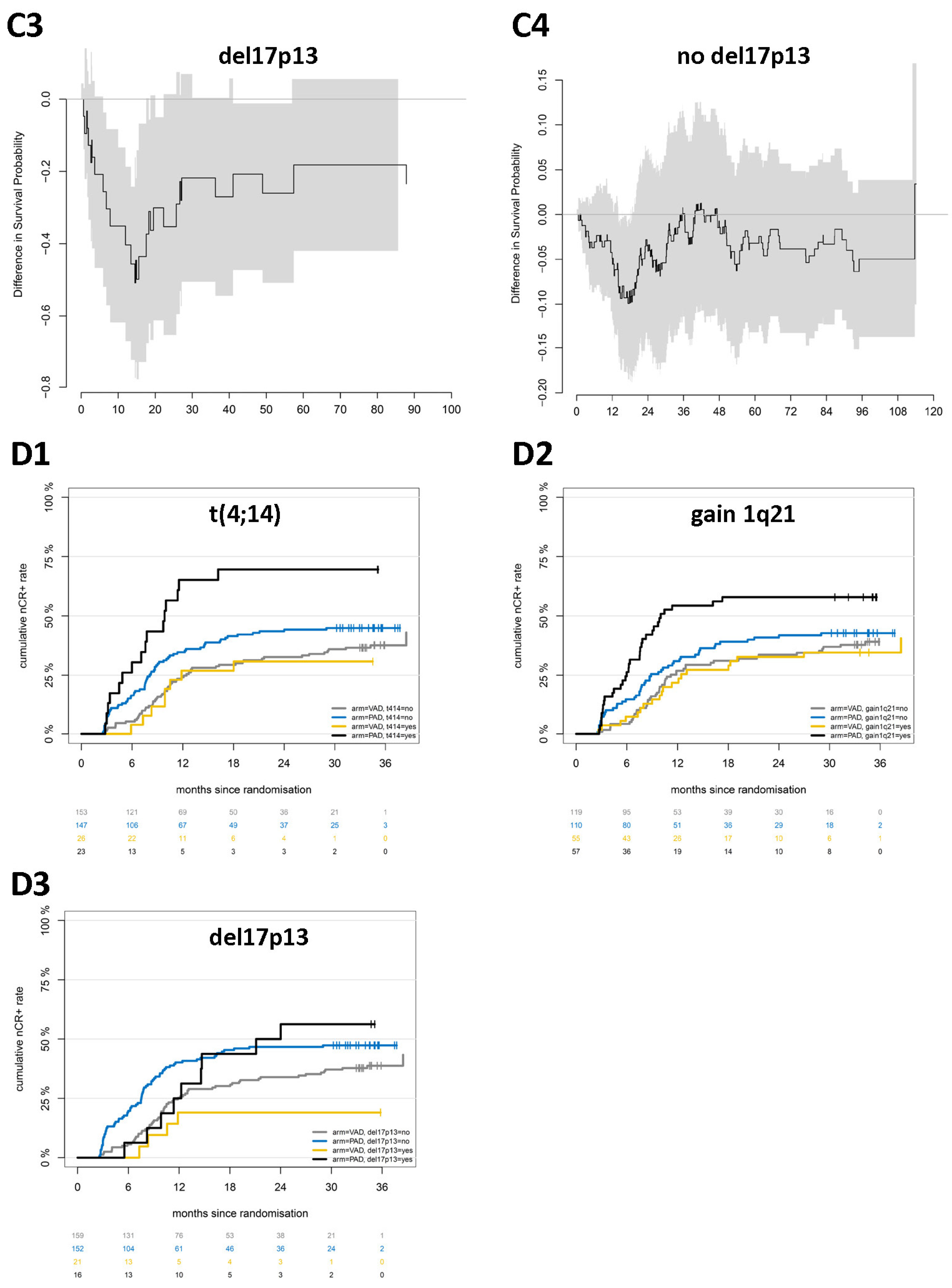
2.3. Responses Achieved by Different Treatment Regimens Are Not Equal
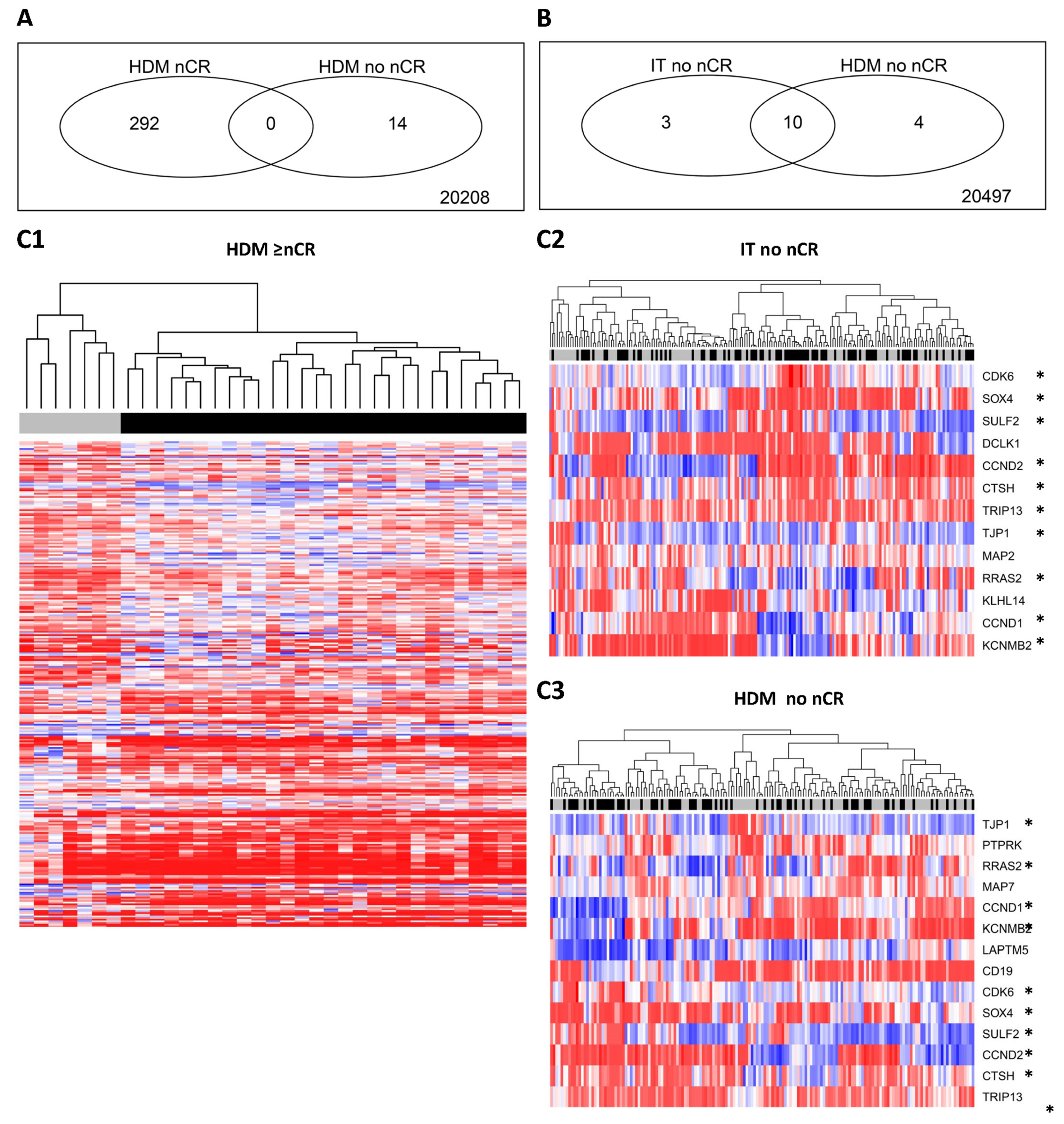
2.4. Molecular Background—Specific Gene Expression Patterns for Patients Responding to PAD- vs. VAD-Based Induction
2.5. Who Benefits from the Inclusion of Bortezomib in Upfront Treatment?
3. Discussion
3.1. Chromosomal Aberrations Determine Long-Term Prognosis by Different Patterns of Association with Proliferation and Treatment-Dependent Response
3.2. Responses Achieved by Different Treatment Regimens Are Not Equal
3.3. Which Patients Profit from Bortezomib-Based Upfront Treatment, and What Lessons Might Be Drawn for Situations of Economic Constraints?
4. Methods
4.1. Study Design and Participants
4.2. Purification of CD138+ Plasma Cells
4.3. iFISH Analysis
4.4. Gene Expression Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Ludwig, H.; Kainz, S.; Schreder, M.; Zojer, N.; Hinke, A. SLiM CRAB criteria revisited: Temporal trends in prognosis of patients with smoldering multiple myeloma who meet the definition of ‘biomarker-defined early multiple myeloma’—A systematic review with meta-analysis. EClinicalMedicine 2023, 58, 101910. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Baker, R.; Martinez, G.; Min, C.-K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; Leblanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Moreau, P.; Kumar, S.K.; San Miguel, J.; Davies, F.; Zamagni, E.; Bahlis, N.; Ludwig, H.; Mikhael, J.; Terpos, E.; Schjesvold, F.; et al. Treatment of relapsed and refractory multiple myeloma: Recommendations from the International Myeloma Working Group. Lancet Oncol. 2021, 22, e105–e118. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up(dagger). Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Hayman, S.R.; Lacy, M.Q.; Dispenzieri, A.; Geyer, S.M.; Kabat, B.; Zeldenrust, S.R.; Kumar, S.; Greipp, P.R.; Fonseca, R.; et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005, 106, 4050–4053. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Stewart, A.K.; Vallone, M.; Molineaux, C.J.; Kunkel, L.A.; Gerecitano, J.F.; Orlowski, R.Z. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 2009, 15, 7085–7091. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Roy, V.; Reeder, C.B.; Lacy, M.Q.; Gertz, A.M.; Laumann, K.; Thompson, A.M.; Witzig, E.T.; Buadi, F.K.; et al. Phase 2 trial of ixazomib in patients with relapsed multiple myeloma not refractory to bortezomib. Blood Cancer J. 2015, 5, e338. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Weller, E.; Hideshima, T.; Mitsiades, C.; Davies, F.; LeBlanc, R.; Catley, L.P.; Doss, D.; Kelly, K.; et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 2002, 100, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Schey, S.; Fields, P.; Bartlett, J.; Clarke, I.; Ashan, G.; Knight, R.; Streetly, M.; Dalgleish, A. Phase I study of an immunomodulatory thalidomide analog, CC-4047, in relapsed or refractory multiple myeloma. J. Clin. Oncol. 2004, 22, 3269–3276. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-cell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): A multicentre, open-label, single-arm, phase 1 study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Wu, J.F.; Dhakal, B. BCMA-targeted CAR-T cell therapies in relapsed and/or refractory multiple myeloma: Latest updates from 2023 ASCO Annual Meeting. J. Hematol. Oncol. 2023, 16, 86. [Google Scholar] [CrossRef]
- Chari, A.; Berdeja, J.G.; Oriol, A.; Van De Donk, N.W.C.J.; Rodriguez, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Verona, R.; Girgis, S.; et al. A Phase 1, First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) x CD3 Bispecific Antibody, in Patients with Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136 (Suppl. S1), 40–41. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.; Rodriguez-Otero, P.; Askari, E.; Mateos, M.-V.; Costa, L.J.; Verona, R.I.; et al. Updated Phase 1 Results from MonumenTAL-1: First-in-Human Study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D × CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2021, 138 (Suppl. S1), 158. [Google Scholar] [CrossRef]
- Mailankody, S.; Diamonte, C.; Fitzgerald, L.; Kane, P.; Wang, X.; Sikder, D.S.; Senechal, B.; Bermudez, V.P.; Frias, D.; Morgan, J.; et al. Phase I First-in-Class Trial of MCARH109, a G Protein Coupled Receptor Class C Group 5 Member D (GPRC5D) Targeted CAR T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2021, 138 (Suppl. S1), 827. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. S1), 42–43. [Google Scholar] [CrossRef]
- Trudel, S.; Cohen, A.D.; Krishnan, A.Y.; Fonseca, R.; Spencer, A.; Berdeja, J.G.; Lesokhin, A.; Forsberg, A.P.; Laubach, J.P.; Costa, L.J.; et al. Cevostamab Monotherapy Continues to Show Clinically Meaningful Activity and Manageable Safety in Patients with Heavily Pre-Treated Relapsed/Refractory Multiple Myeloma (RRMM): Updated Results from an Ongoing Phase I Study. Blood 2021, 138 (Suppl. S1), 157. [Google Scholar] [CrossRef]
- Zhao, J.; Ren, Q.; Liu, X.; Guo, X.; Song, Y. Bispecific antibodies targeting BCMA, GPRC5D, and FcRH5 for multiple myeloma therapy: Latest updates from ASCO 2023 Annual Meeting. J. Hematol. Oncol. 2023, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; Grant, S.J.; Wildes, T.M. Chimeric Antigen Receptor T-Cell and Bispecific Antibody Therapy in Multiple Myeloma: Moving into the Future. J. Clin. Oncol. 2023, 41, 4416–4429. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Galisteo, A.; Álvarez-Vallina, L.; Sanz, L. Bi- and trispecific immune cell engagers for immunotherapy of hematological malignancies. J. Hematol. Oncol. 2023, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Neri, P.; Bahlis, N.J. BCMA- or GPRC5D-targeting bispecific antibodies in multiple myeloma: Efficacy, safety, and resistance mechanisms. Blood 2024, 143, 1211–1217. [Google Scholar] [CrossRef]
- Sonneveld, P.; Schmidt-Wolf, I.G.; van der Holt, B.; El Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Vellenga, E.; Broyl, A.; Blau, I.W.; et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J. Clin. Oncol. 2012, 30, 2946–2955. [Google Scholar] [CrossRef]
- Neben, K.; Lokhorst, H.M.; Jauch, A.; Bertsch, U.; Hielscher, T.; van der Holt, B.; Salwender, H.; Blau, I.W.; Weisel, K.; Pfreundschuh, M.; et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood 2012, 119, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, H.; Lokhorst, H.M.; Mai, E.K.; van der Holt, B.; Blau, I.W.; Zweegman, S.; Weisel, K.C.; Vellenga, E.; Pfreundschuh, M.; Kersten, M.J.; et al. Bortezomib before and after high-dose therapy in myeloma: Long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 2018, 32, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Scheid, C.; Sonneveld, P.; Schmidt-Wolf, I.G.; van der Holt, B.; el Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Blau, I.W.; Vellenga, E.; et al. Bortezomib before and after autologous stem cell transplantation overcomes the negative prognostic impact of renal impairment in newly diagnosed multiple myeloma: A subgroup analysis from the HOVON-65/GMMG-HD4 trial. Haematologica 2014, 99, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Hose, D.; Rème, T.; Hielscher, T.; Moreaux, J.; Messner, T.; Seckinger, A.; Benner, A.; Shaughnessy, J.D.; Barlogie, B.; Zhou, Y.; et al. Proliferation is a central independent prognostic factor and target for personalized and risk adapted treatment in multiple myeloma. Haematologica 2011, 96, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Van Wier, A.S.; Chng, W.J.; Ketterling, R.; Lacy, M.Q.; Dispenzieri, A.; Bergsagel, P.L.; Rajkumar, S.V.; Greipp, P.R.; Litzow, M.R.; et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006, 20, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Hegenbart, U.; Kunz, C.; Granzow, M.; Benner, A.; Seckinger, A.; Kimmich, C.; Goldschmidt, H.; Ho, A.D.; Hose, D.; et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J. Clin. Oncol. 2015, 33, 1371–1378. [Google Scholar] [CrossRef]
- Chng, W.J.; Dispenzieri, A.; Chim, C.-S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; San-Miguel, J.F.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- D’Agostino, M.; Cairns, D.A.; Lahuerta, J.J.; Wester, R.; Bertsch, U.; Waage, A.; Zamagni, E.; Mateos, M.-V.; Dall’Olio, D.; van de Donk, N.W.; et al. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report Within the HARMONY Project. J. Clin. Oncol. 2022, 40, 3406–3418. [Google Scholar] [CrossRef]
- Abdallah, N.H.; Binder, M.; Rajkumar, S.V.; Greipp, P.T.; Kapoor, P.; Dispenzieri, A.; Gertz, M.A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; et al. A simple additive staging system for newly diagnosed multiple myeloma. Blood Cancer J. 2022, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Mateos, M.-V.; Gutierrez, N.C.; Rajkumar, S.V.; Miguel, J.F.S. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013, 121, 884–892. [Google Scholar] [CrossRef]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.-J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef]
- Durie, B.G.; Salmon, S.E. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975, 36, 842–854. [Google Scholar] [CrossRef]
- Durie, B.G.M.; Harousseau, J.-L.; Miguel, J.S.; Blade, J.; Barlogie, B.; Anderson, K.; Gertz, M.; Dimopoulos, M.; Westin, J.; Sonneveld, P.; et al. International uniform response criteria for multiple myeloma. Leukemia 2006, 20, 1467–1473. [Google Scholar] [CrossRef]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef]
- Hose, D.; Beck, S.; Salwender, H.; Emde, M.; Bertsch, U.; Kunz, C.; Scheid, C.; Hänel, M.; Weisel, K.; Hielscher, T.; et al. Prospective target assessment and multimodal prediction of survival for personalized and risk-adapted treatment strategies in multiple myeloma in the GMMG-MM5 multicenter trial. J. Hematol. Oncol. 2019, 12, 65. [Google Scholar] [CrossRef]
- Emde-Rajaratnam, M.; Beck, S.; Benes, V.; Salwender, H.; Bertsch, U.; Scheid, C.; Hänel, M.; Weisel, K.; Hielscher, T.; Raab, M.S.; et al. RNA-sequencing based first choice of treatment and determination of risk in multiple myeloma. Front. Immunol. 2023, 14, 1286700. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Wuilleme, S.; Robillard, N.; Lode, L.; Magrangeas, F.; Beris, H.; Harousseau, J.L.; Proffitt, J.; Minvielle, S.; Avet-Loiseau, H. Ploidy, as detected by fluorescence in situ hybridization, defines different subgroups in multiple myeloma. Leukemia 2005, 19, 275–278. [Google Scholar] [CrossRef]
- Meißner, T.; Seckinger, A.; Rème, T.; Hielscher, T.; Möhler, T.; Neben, K.; Goldschmidt, H.; Klein, B.; Hose, D. Gene Expression Profiling in Multiple Myeloma—Reporting of Entities, Risk, and Targets in Clinical Routine. Clin. Cancer Res. 2011, 17, 7240–7247. [Google Scholar] [CrossRef]
- Hose, D.; Rème, T.; Meissner, T.; Moreaux, J.; Seckinger, A.; Lewis, J.; Benes, V.; Benner, A.; Hundemer, M.; Hielscher, T.; et al. Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood 2009, 113, 4331–4340. [Google Scholar] [CrossRef]
- Gray, R.J. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann. Stat. 1988, 16, 1141–1154. [Google Scholar] [CrossRef]
- Wu, Z.; Irizarry, R.A.; Gentleman, R.; Martinez-Murillo, F.; Spencer, F. A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. J. Am. Stat. Assoc. 2004, 99, 909–917. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R.; et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Greipp, P.R.; Miguel, J.S.; Durie, B.G.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Mai, E.K.; Bertsch, U.; Dürig, J.; Kunz, C.; Haenel, M.; Blau, I.W.; Munder, M.; Jauch, A.; Schurich, B.; Hielscher, T.; et al. Phase III trial of bortezomib, cyclophosphamide and dexamethasone (VCD) versus bortezomib, doxorubicin and dexamethasone (PAd) in newly diagnosed myeloma. Leukemia 2015, 29, 1721–1729. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pts. [%] | Proli- Feration | Tumor Mass | ≥nCR after HDM | CREA ≥2 | PFS | OS | Benefit Shown | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALL | VAD | PAD | ALL | VAD | PAD | ALL | VAD | PAD | ||||||
| GPI | ∕ | ∕ | = | = | = | = | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | yes | |
| t(4;14) | 13.9 | ↑ | ↑ | = | = | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | yes |
| gain 1q21 | 32.6 | ↑ | = | ↑ | = | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | yes |
| del17p13 | 10.6 | ↑ | = | ↓ | = | ↓ | ↑ | ↓ | ↓ | = | ↓ | ↓ | = | yes |
| t(11;14) | 19.2 | = | = | = | = | ↓ | = | = | = | = | = | = | = | yes |
| del13q14 only | 38.8 | ↑ | = | = | = | = | = | = | = | = | ↓ | ↓ | ↓ | no |
| hyperdiploidy | 51.3 | ↓ | = | = | = | = | = | = | = | = | = | = | = | yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seckinger, A.; Salwender, H.; Martin, H.; Scheid, C.; Hielscher, T.; Bertsch, U.; Hummel, M.; Jauch, A.; Knauf, W.; Emde-Rajaratnam, M.; et al. Molecular Long-Term Analysis of the GMMG-HD4 Trial in Multiple Myeloma—Patterns of Association of Chromosomal Aberrations with Response and Proliferation Determining Survival in Selecting Treatments in View of Limited Resources in Low- and Middle-Income Countries. Int. J. Mol. Sci. 2024, 25, 6431. https://doi.org/10.3390/ijms25126431
Seckinger A, Salwender H, Martin H, Scheid C, Hielscher T, Bertsch U, Hummel M, Jauch A, Knauf W, Emde-Rajaratnam M, et al. Molecular Long-Term Analysis of the GMMG-HD4 Trial in Multiple Myeloma—Patterns of Association of Chromosomal Aberrations with Response and Proliferation Determining Survival in Selecting Treatments in View of Limited Resources in Low- and Middle-Income Countries. International Journal of Molecular Sciences. 2024; 25(12):6431. https://doi.org/10.3390/ijms25126431
Chicago/Turabian StyleSeckinger, Anja, Hans Salwender, Hans Martin, Christof Scheid, Thomas Hielscher, Uta Bertsch, Manuela Hummel, Anna Jauch, Wolfgang Knauf, Martina Emde-Rajaratnam, and et al. 2024. "Molecular Long-Term Analysis of the GMMG-HD4 Trial in Multiple Myeloma—Patterns of Association of Chromosomal Aberrations with Response and Proliferation Determining Survival in Selecting Treatments in View of Limited Resources in Low- and Middle-Income Countries" International Journal of Molecular Sciences 25, no. 12: 6431. https://doi.org/10.3390/ijms25126431
APA StyleSeckinger, A., Salwender, H., Martin, H., Scheid, C., Hielscher, T., Bertsch, U., Hummel, M., Jauch, A., Knauf, W., Emde-Rajaratnam, M., Beck, S., Neben, K., Dührig, J., Lindemann, W., Schmidt-Wolf, I. G. H., Hänel, M., Blau, I. W., Weisel, K., Weinhold, N., ... Hose, D. (2024). Molecular Long-Term Analysis of the GMMG-HD4 Trial in Multiple Myeloma—Patterns of Association of Chromosomal Aberrations with Response and Proliferation Determining Survival in Selecting Treatments in View of Limited Resources in Low- and Middle-Income Countries. International Journal of Molecular Sciences, 25(12), 6431. https://doi.org/10.3390/ijms25126431