
IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells


Abstract
1. Introduction
2. Results
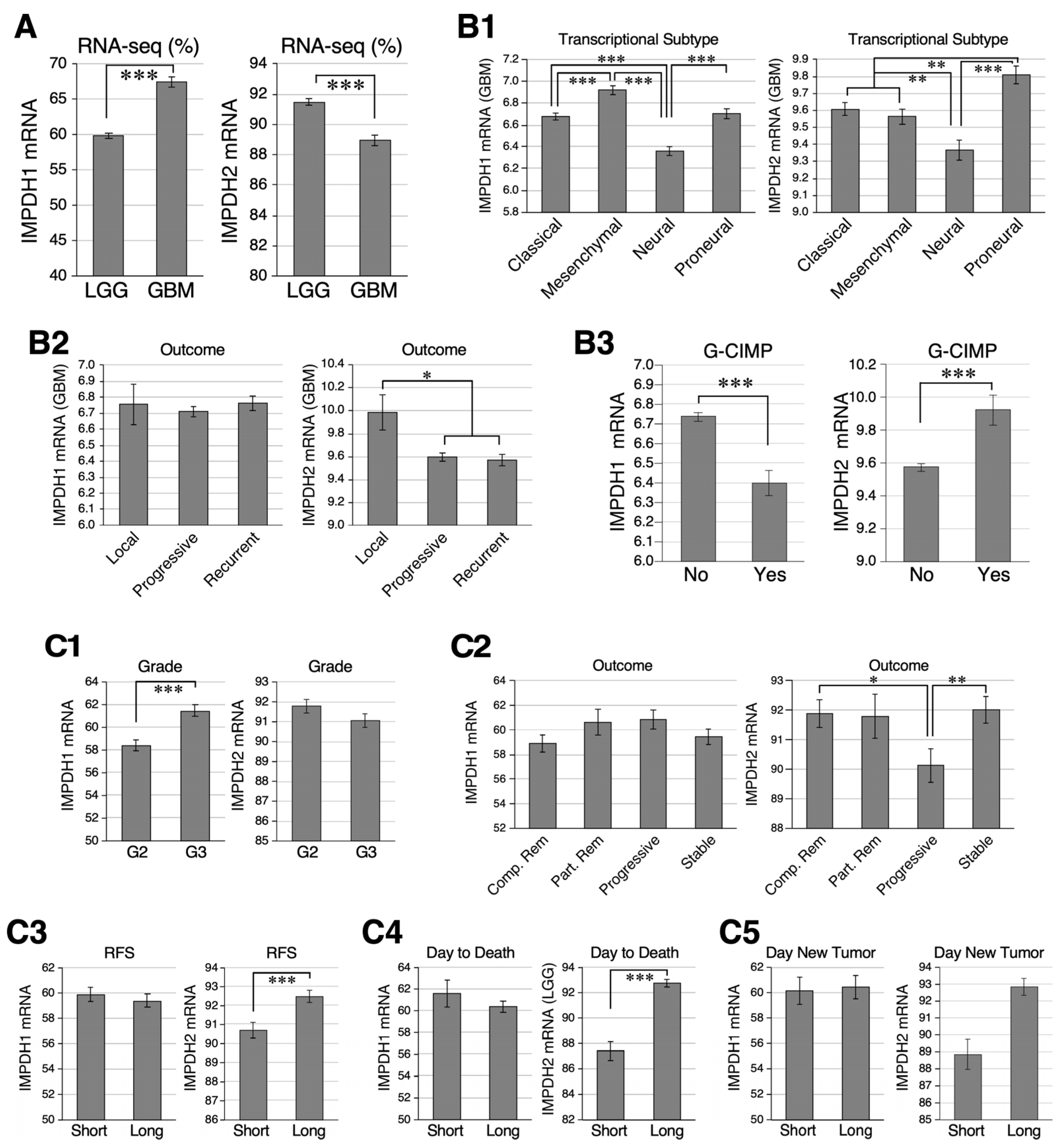
2.1. Relative Abundance of IMPDH1 and IMPDH2 in GBM and LGG Samples
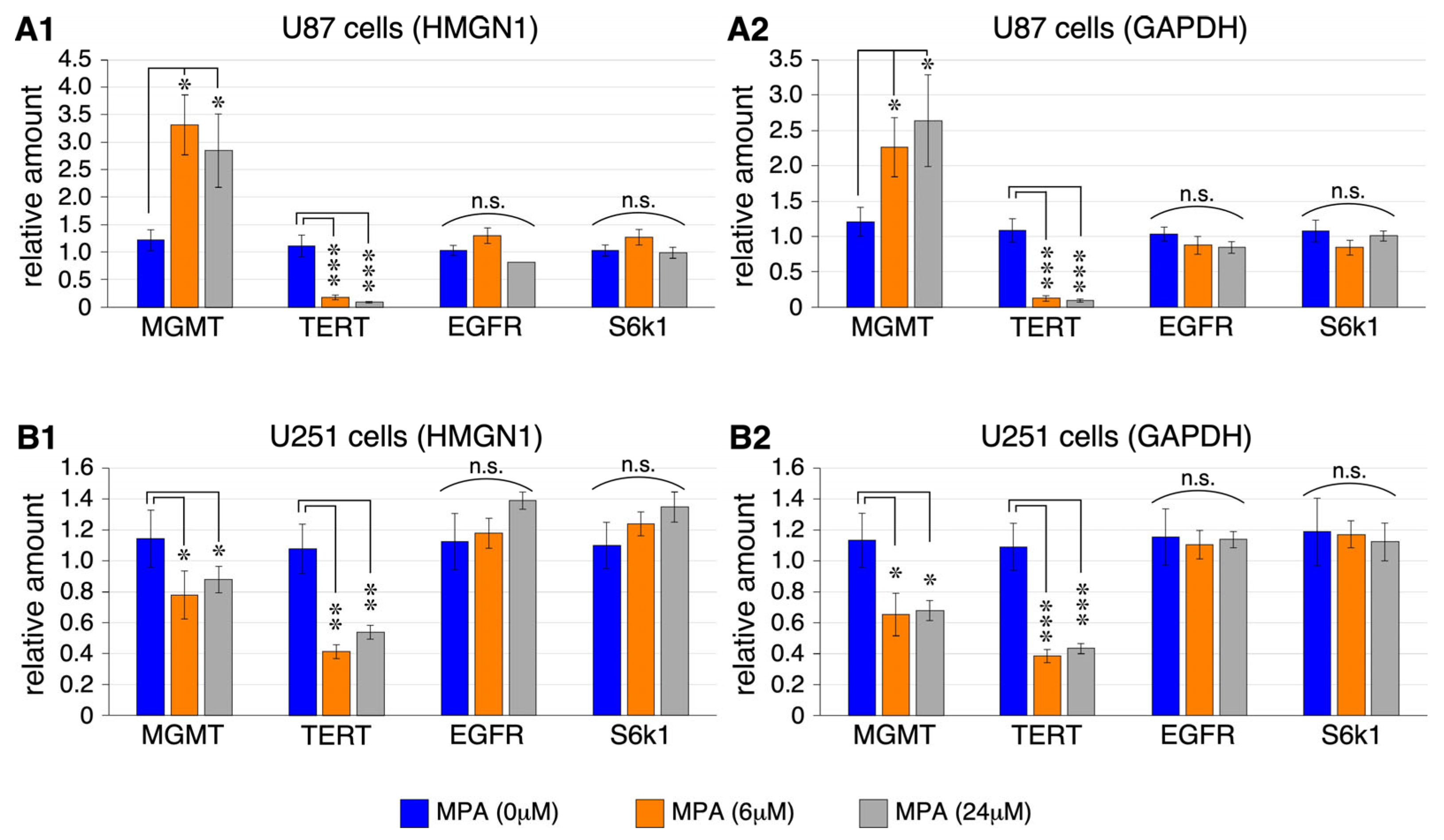
2.2. MPA Effect on the Expression of MGMT, TERT, EGFR, and S6k1 in U87 and U251 Cells
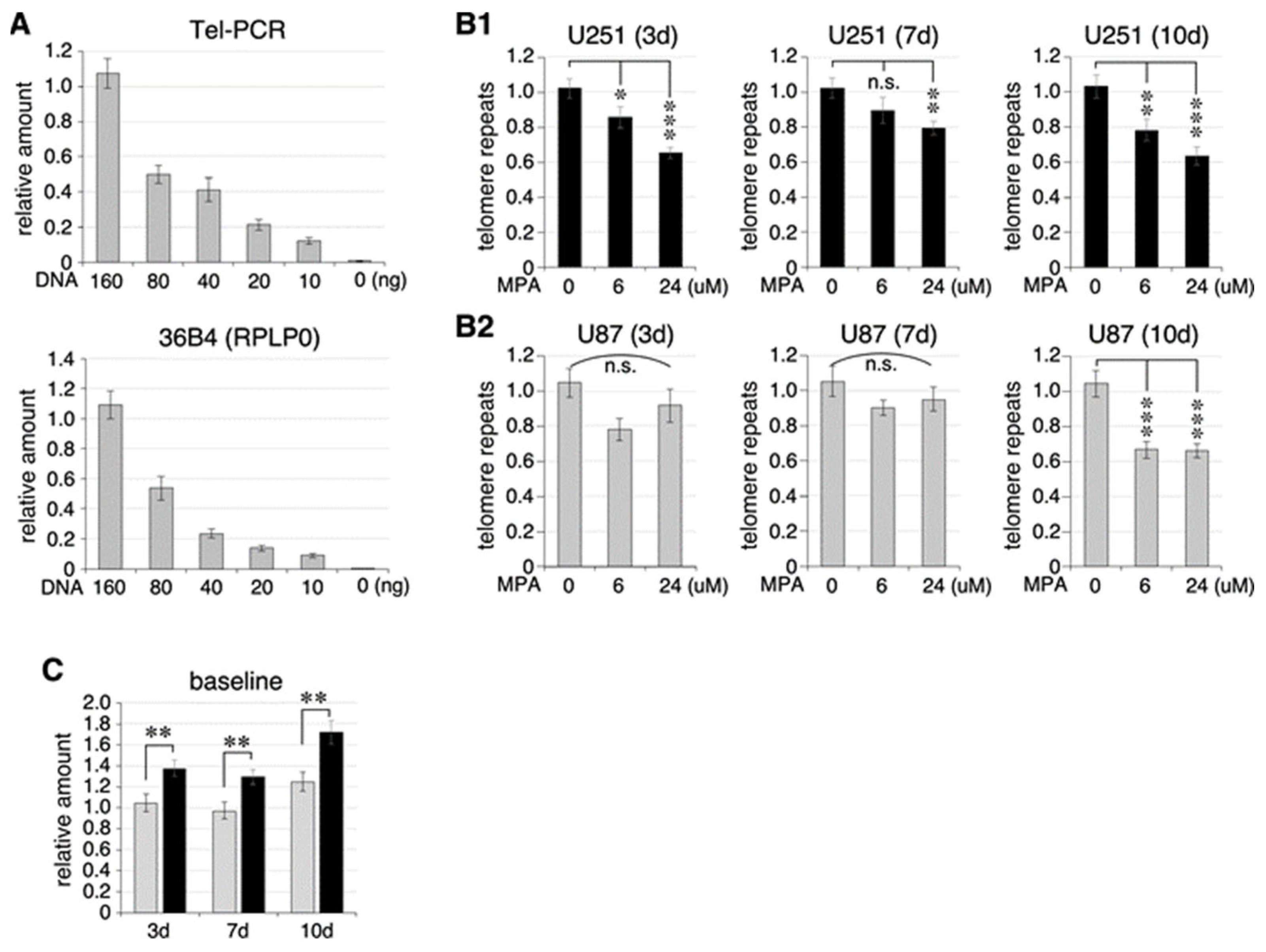
2.3. Effect of MPA Treatment on Telomere Length in U87 and U251 Cells
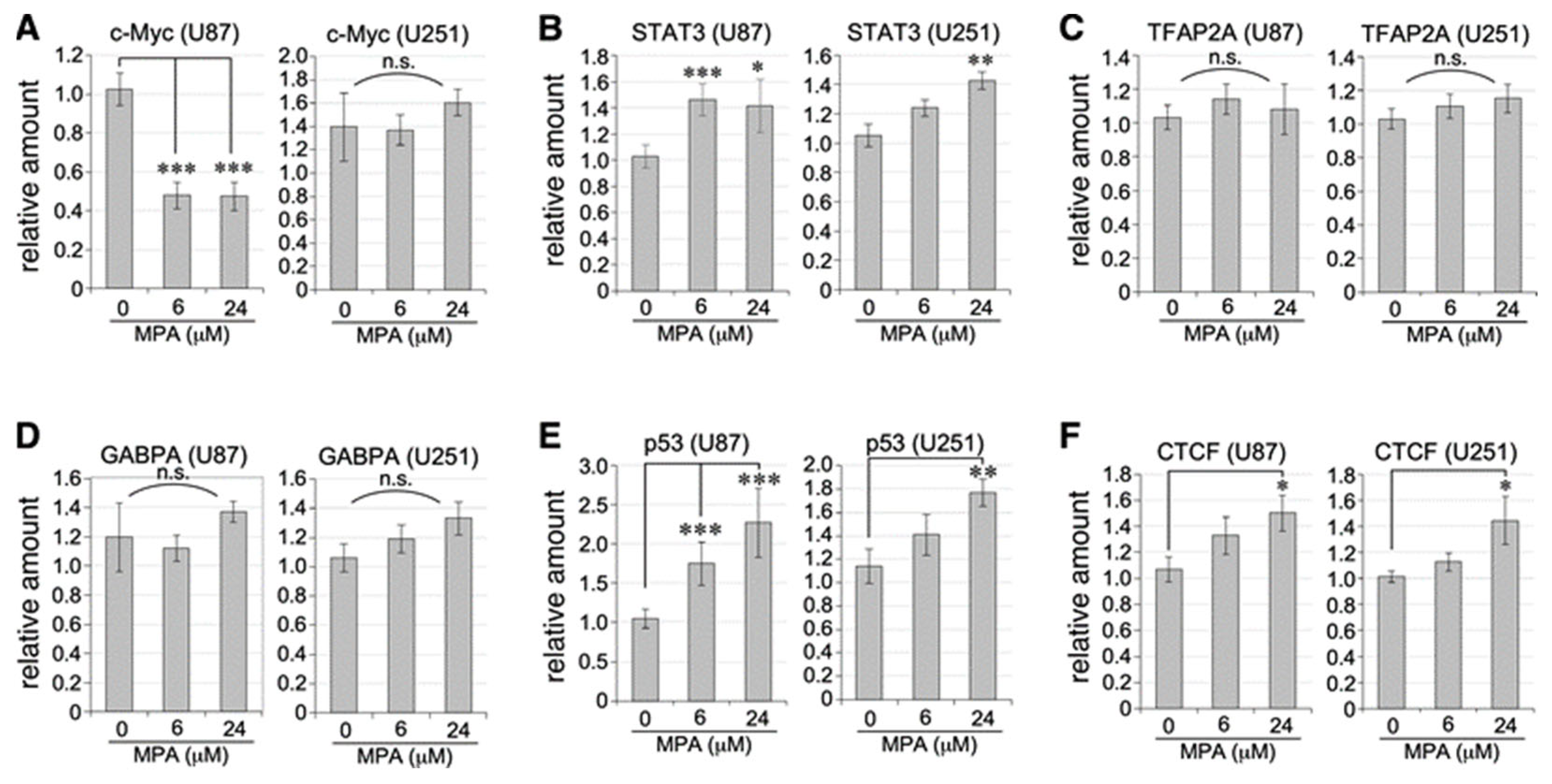
2.4. Transcriptional Regulation of TERT Activators and Repressors by MPA Treatment
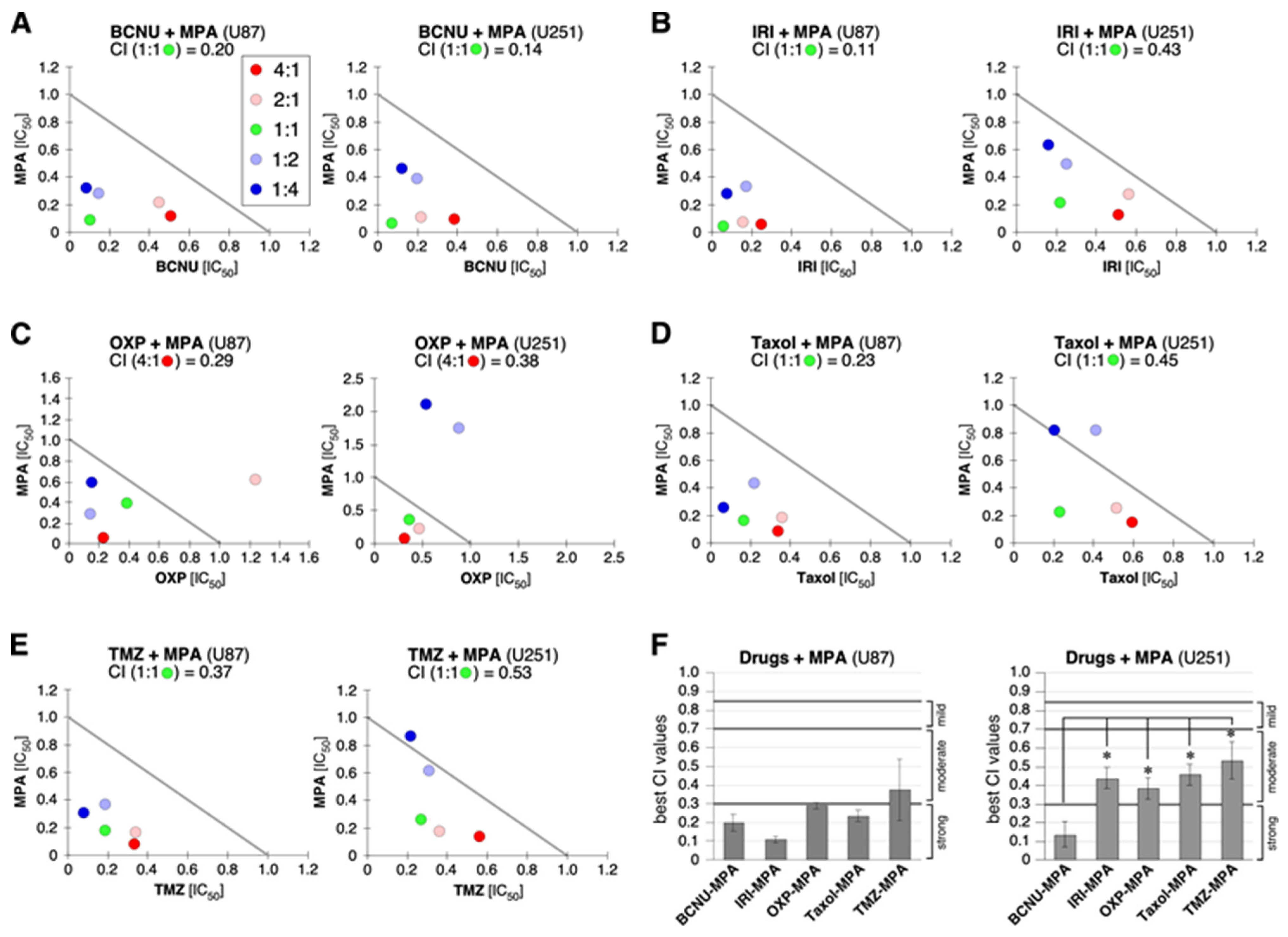
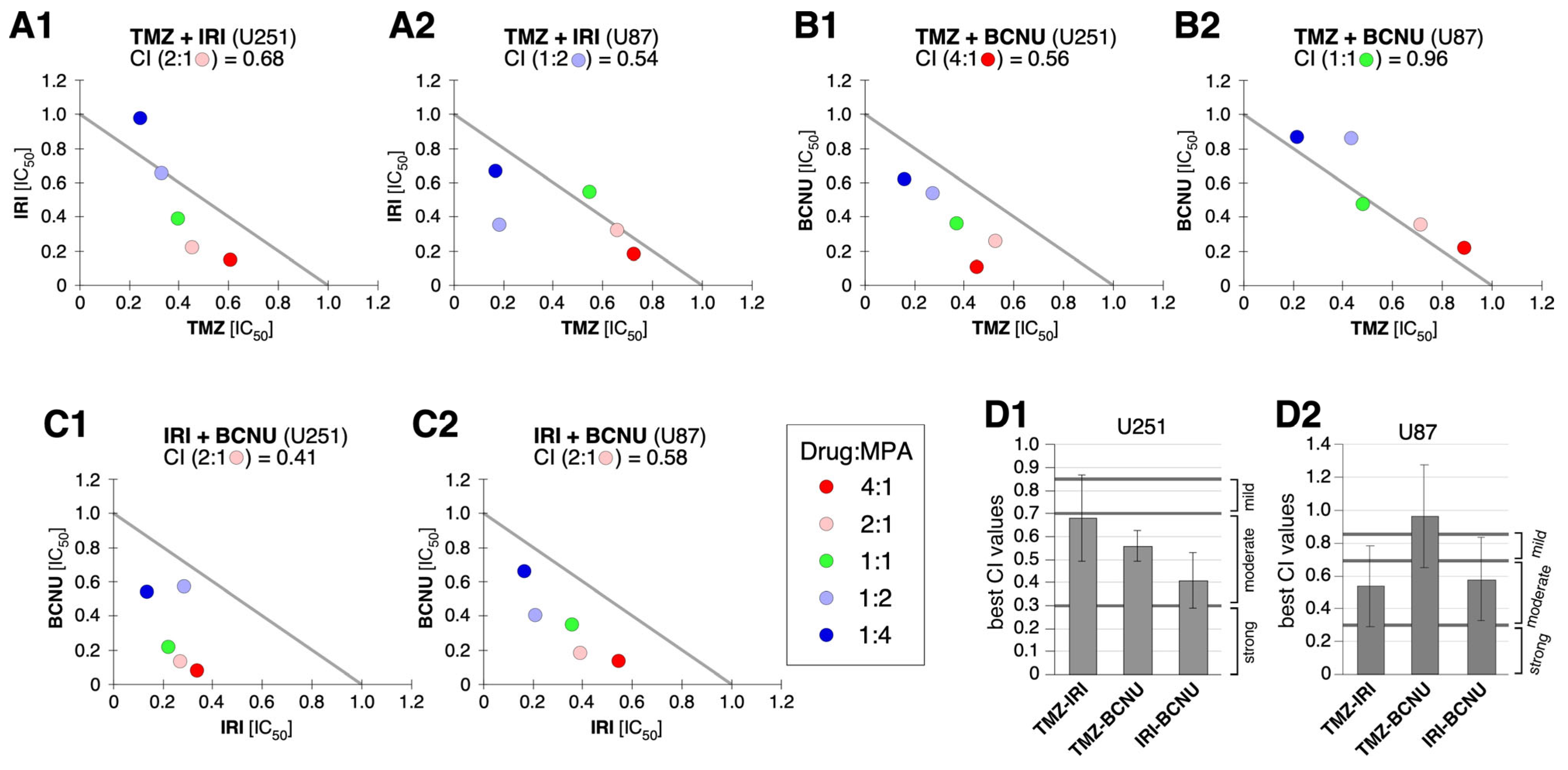
2.5. Synergy between MPA and BCNU and Other Drugs
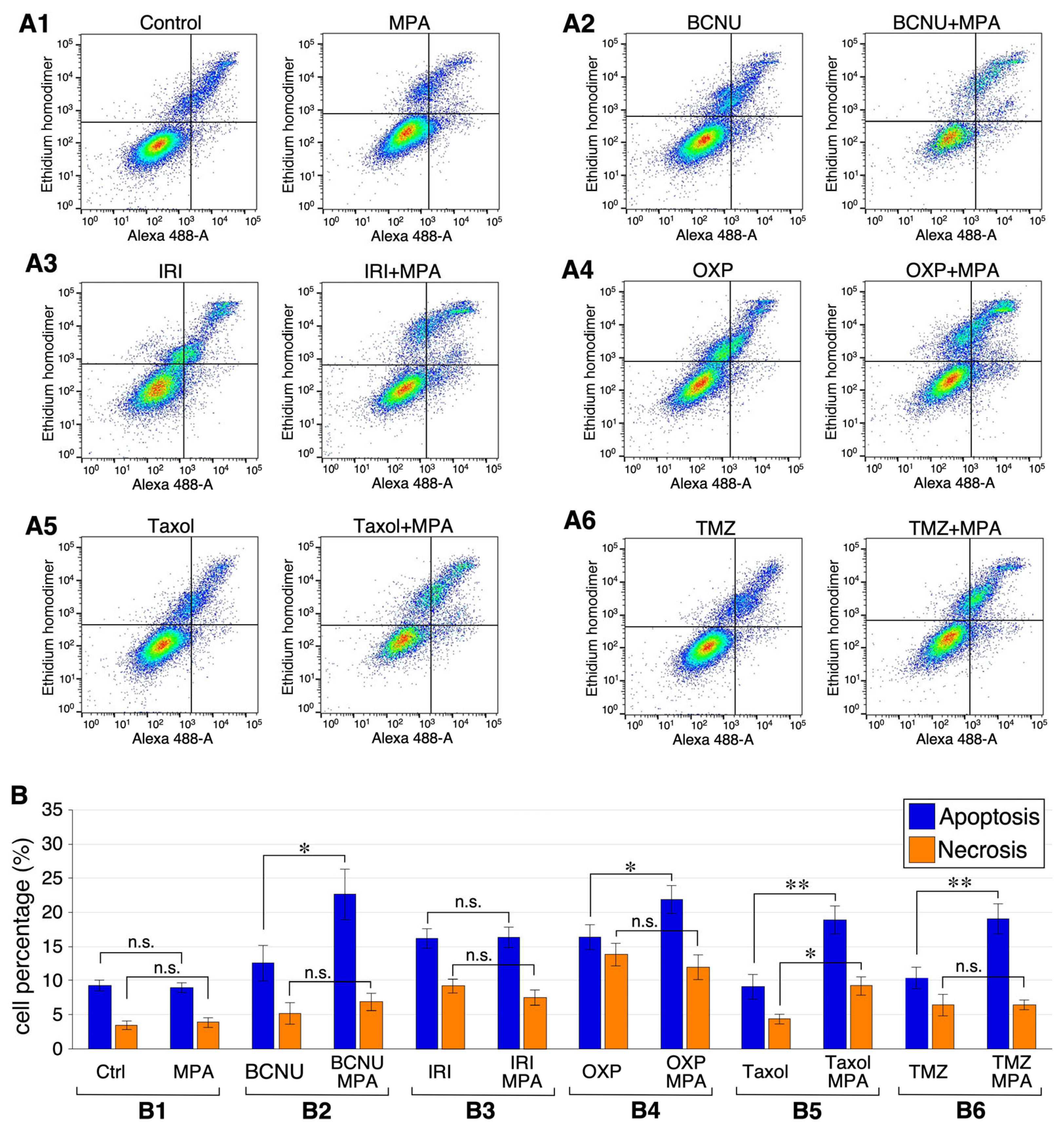
2.6. Combining MPA with BCNU, OXP, Taxol, or TMZ Increases Apoptosis in U251 Cells
3. Discussion
3.1. IMPDH Inhibition Downregulates TERT Expression
3.2. Cytotoxic Synergy between MPA and Anti-GBM Agents
3.3. Limitations and Challenges Ahead
4. Materials and Methods
4.1. TCGA Analysis
4.2. Cell Culture
4.3. Quantitative (Real-Time) RT-PCR Assay
4.4. qPCR-Based Telomere Length Assay
4.5. Thiazolyl Blue Tetrazolium Bromide (MTT) Assay
4.6. Combination Index (CI) and Isobologram
4.7. Apoptosis and Necrosis Measurement
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Silvani, A.; Gaviani, P.; Lamperti, E.A.; Eoli, M.; Falcone, C.; Dimeco, F.; Milanesi, I.M.; Erbetta, A.; Boiardi, A.; Fariselli, L.; et al. Cisplatinum and BCNU chemotherapy in primary glioblastoma patients. J. Neurooncol. 2009, 94, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, B.M.; Esrailian, E.; Laine, L.; Chamberlain, M.C. Clinical impact of adjuvant chemotherapy in glioblastoma multiforme: A meta-analysis. CNS Drugs 2007, 21, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018. Neuro Oncol. 2021, 23 (Suppl. S2), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Sintchak, M.D.; Fleming, M.A.; Futer, O.; Raybuck, S.A.; Chambers, S.P.; Caron, P.R.; Murcko, M.A.; Wilson, K.P. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 1996, 85, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Savchuk, S.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 2021, 144, 1230–1246. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.P.; Kharkar, P.S. Discovery of novel human inosine 5’-monophosphate dehydrogenase 2 (hIMPDH2) inhibitors as potential anticancer agents. Eur. J. Med. Chem. 2018, 158, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.P.; Kharkar, P.S. Newer human inosine 5’-monophosphate dehydrogenase 2 (hIMPDH2) inhibitors as potential anticancer agents. J. Enzym. Inhib. Med. Chem. 2018, 33, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Stojkovic, T.; de Seze, J. Mycophenolate mofetil and neurological diseases. Lupus 2005, 14 (Suppl. S1), s42–s45. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; Dai, M.S.; Lu, H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J. Biol. Chem. 2008, 283, 12387–12392. [Google Scholar] [CrossRef] [PubMed]
- Heinschink, A.; Raab, M.; Daxecker, H.; Griesmacher, A.; Muller, M.M. In vitro effects of mycophenolic acid on cell cycle and activation of human lymphocytes. Clin. Chim. Acta 2000, 300, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, J.; Yee, A.; Xiong, Y.; Mitchell, B.S. Effects of guanine nucleotide depletion on cell cycle progression in human T lymphocytes. Blood 1998, 91, 2896–2904. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Gathy, K.; Santiago, L.; Chen, E.; Huang, M.; Graves, L.M.; Mitchell, B.S. Induction of apoptosis in IL-3-dependent hematopoietic cell lines by guanine nucleotide depletion. Blood 2003, 101, 4958–4965. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Cheng, X.; Fandy, T.E.; Srivastava, R.K.; Wu, S.; Shankar, S.; Bauer, K.; Shaughnessy, J.; Tricot, G. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol. Cancer Ther. 2006, 5, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hideshima, T.; Hamasaki, M.; Raje, N.; Kumar, S.; Podar, K.; Le Gouill, S.; Shiraishi, N.; Yasui, H.; Roccaro, A.M.; et al. Novel inosine monophosphate dehydrogenase inhibitor VX-944 induces apoptosis in multiple myeloma cells primarily via caspase-independent AIF/Endo G pathway. Oncogene 2005, 24, 5888–5896. [Google Scholar] [CrossRef]
- Huang, M.; Itahana, K.; Zhang, Y.; Mitchell, B.S. Depletion of guanine nucleotides leads to the Mdm2-dependent proteasomal degradation of nucleostemin. Cancer Res. 2009, 69, 3004–3012. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M. Mycophenolic acid and its mechanism of action in cancer and psoriasis. Jpn. J. Antibiot. 1977, 30, 85092. [Google Scholar]
- Behrend, M. Adverse gastrointestinal effects of mycophenolate mofetil: Aetiology, incidence and management. Drug Saf. 2001, 24, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Santiago, L.; Mitchell, B.S. Synergy between imatinib and mycophenolic acid in inducing apoptosis in cell lines expressing Bcr-Abl. Blood 2005, 105, 3270–3277. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Meng, L.; Tsai, R.Y. GTP depletion synergizes the anti-proliferative activity of chemotherapeutic agents in a cell type-dependent manner. Biochem. Biophys. Res. Commun. 2011, 414, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Q.; Wang, Y.; Crawford, M.; Bhupal, P.K.; Gao, X.; Xie, H.; Liang, D.; Cheng, Y.L.; Liu, X.; et al. Designing a Mucoadhesive ChemoPatch to Ablate Oral Dysplasia for Cancer Prevention. Small 2022, 18, e2201561. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, G.; Efimov, V.; Raevskiy, M.; Rumiantsev, P.; Gudkov, A.; Belogurova-Ovchinnikova, O.; Sorokin, M.; Buzdin, A. Reclassification of TCGA Diffuse Glioma Profiles Linked to Transcriptomic, Epigenetic, Genomic and Clinical Data, According to the 2021 WHO CNS Tumor Classification. Int. J. Mol. Sci. 2022, 24, 157. [Google Scholar] [CrossRef]
- Karsy, M.; Guan, J.; Cohen, A.L.; Jensen, R.L.; Colman, H. New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr. Neurol. Neurosci. Rep. 2017, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Avin, B.A.; Wang, Y.; Gilpatrick, T.; Workman, R.E.; Lee, I.; Timp, W.; Umbricht, C.B.; Zeiger, M.A. Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosomes Cancer 2019, 58, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.C.; Hung, J.J. Functional role of post-translational modifications of Sp1 in tumorigenesis. J. Biomed. Sci. 2012, 19, 94. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.G.; Jayachandran, G.; Wu, G.; Xu, K.; Roth, J.A.; Ji, L. Tumor-specific activation of human telomerase reverses transcriptase promoter activity by activating enhancer-binding protein-2beta in human lung cancer cells. J. Biol. Chem. 2007, 282, 26460–26470. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhou, J.; Wang, M.; Cong, Y.S. Implications of telomere-independent activities of telomerase reverse transcriptase in human cancer. FEBS J. 2013, 280, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Dratwa, M.; Wysoczanska, B.; Lacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT-Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11, 589929. [Google Scholar] [CrossRef] [PubMed]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.B.; Wu, Y.R.; Liu, C.H.; Hu, C.J.; Yang, S.M.; Li, X.Z. Diverse regulatory manners of human telomerase reverse transcriptase. Cell Commun. Signal. 2019, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Konnikova, L.; Simeone, M.C.; Kruger, M.M.; Kotecki, M.; Cochran, B.H. Signal transducer and activator of transcription 3 (STAT3) regulates human telomerase reverse transcriptase (hTERT) expression in human cancer and primary cells. Cancer Res. 2005, 65, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, X.; Ge, Z.; Jalink, M.; Kyo, S.; Bjorkholm, M.; Gruber, A.; Sjoberg, J.; Xu, D. The telomerase reverse transcriptase (hTERT) gene is a direct target of the histone methyltransferase SMYD3. Cancer Res. 2007, 67, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarria-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef]
- Wei, C.W.; Lin, C.C.; Yu, Y.L.; Lin, C.Y.; Lin, P.C.; Wu, M.T.; Chen, C.J.; Chang, W.; Lin, S.Z.; Chen, Y.L.; et al. n-Butylidenephthalide induced apoptosis in the A549 human lung adenocarcinoma cell line by coupled down-regulation of AP-2alpha and telomerase activity. Acta Pharmacol. Sin. 2009, 30, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Suzuki, H.; Nakajo, Y.; Nakano, A.; Kato, M. Regulation of c-MYC transcriptional activity by transforming growth factor-beta 1-stimulated clone 22. Cancer Sci. 2018, 109, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Xavier-Magalhaes, A.; Woods, W.S.; Nguyen, K.T.; Amen, A.M.; Hayes, J.L.; Fellmann, C.; Gapinske, M.; McKinney, A.M.; Hong, C.; et al. Disruption of the beta1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell 2018, 34, 513–528.e518. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.H.; Jing, X.; Bollenbacher-Reilley, J.; Zhao, D.M.; Haring, J.S.; Yang, B.; Liu, C.; Bishop, G.A.; Harty, J.T.; Leonard, W.J. Targeting the GA binding protein beta1L isoform does not perturb lymphocyte development and function. Mol. Cell Biol. 2008, 28, 4300–4309. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Mu, N.; Wang, N.; Straat, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2019, 38, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Bai, S.; Li, X.; Zhang, Y.; Li, Y.; He, F.; Cheng, W. Establishment and Validation of the Detection of TERT Promoter Mutations by Human Gliomas U251 Cell Lines. Biomed. Res. Int. 2021, 2021, 3271395. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, F.; Jaxel, L.; Lemaire, M.; Turner, J.D.; Perez-Bercoff, D. TERT Promoter Mutations Increase Sense and Antisense Transcription from the TERT Promoter. Biomedicines 2021, 9, 1773. [Google Scholar] [CrossRef] [PubMed]
- Ariyaratana, S.; Loeb, D.M. The role of the Wilms tumour gene (WT1) in normal and malignant haematopoiesis. Expert Rev. Mol. Med. 2007, 9, 1–17. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Peek, G.W.; Tollefsbol, T.O. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 2012, 498, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B. MAD1 and its life as a MYC antagonist: An update. Eur. J. Cell Biol. 2012, 91, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Chan, T.H.; Tollefsbol, T.O. A novel prodrug of epigallocatechin-3-gallate: Differential epigenetic hTERT repression in human breast cancer cells. Cancer Prev. Res. 2011, 4, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Abdullaev, Z.; Guilleret, I.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007, 35, 1245–1256. [Google Scholar] [CrossRef]
- Renaud, S.; Loukinov, D.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005, 33, 6850–6860. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Bjorkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Benjanuwattra, J.; Chaiyawat, P.; Pruksakorn, D.; Koonrungsesomboon, N. Therapeutic potential and molecular mechanisms of mycophenolic acid as an anticancer agent. Eur. J. Pharmacol. 2020, 887, 173580. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Shervington, A.; Patel, R.; Lu, C.; Cruickshanks, N.; Lea, R.; Roberts, G.; Dawson, T.; Shervington, L. Telomerase subunits expression variation between biopsy samples and cell lines derived from malignant glioma. Brain Res. 2007, 1134, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Purkait, S.; Jha, P.; Sharma, M.C.; Suri, V.; Sharma, M.; Kale, S.S.; Sarkar, C. CDKN2A deletion in pediatric versus adult glioblastomas and predictive value of p16 immunohistochemistry. Neuropathology 2013, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; McLendon, R.E.; Kerby, T.; Dugan, M.; Bigner, S.H.; Henry, A.J.; Ashley, D.M.; Krischer, J.; Lovell, S.; Rasheed, K.; et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J. Clin. Oncol. 1998, 16, 3851–3857. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef]
- Weller, M.; Rieger, J.; Grimmel, C.; Van Meir, E.G.; De Tribolet, N.; Krajewski, S.; Reed, J.C.; von Deimling, A.; Dichgans, J. Predicting chemoresistance in human malignant glioma cells: The role of molecular genetic analyses. Int. J. Cancer 1998, 79, 640–644. [Google Scholar] [CrossRef]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef] [PubMed]
- Ponten, J. The 1975 Nobel prize in physiology or medicine: Interaction between genetic cell factors and tumor virus. Lakartidningen 1975, 72, 4264–4267. [Google Scholar] [PubMed]
- Qiang, L.; Yang, Y.; Ma, Y.J.; Chen, F.H.; Zhang, L.B.; Liu, W.; Qi, Q.; Lu, N.; Tao, L.; Wang, X.T.; et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Liau, L.M. IDH mutations in human glioma. Neurosurg. Clin. N. Am. 2012, 23, 471–480. [Google Scholar] [CrossRef]
- Cai, J.; Zhu, P.; Zhang, C.; Li, Q.; Wang, Z.; Li, G.; Wang, G.; Yang, P.; Li, J.; Han, B.; et al. Detection of ATRX and IDH1-R132H immunohistochemistry in the progression of 211 paired gliomas. Oncotarget 2016, 7, 16384–16395. [Google Scholar] [CrossRef] [PubMed]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, D., Jr.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Lin, T.C.; McGrail, D.J.; Bhupal, P.K.; Ku, Y.H.; Zhang, W.; Meng, L.; Lin, S.Y.; Peng, G.; Tsai, R.Y.L. Nucleostemin reveals a dichotomous nature of genome maintenance in mammary tumor progression. Oncogene 2019, 38, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; McGrail, D.J.; Bhupal, P.K.; Zhang, W.; Lin, K.Y.; Ku, Y.H.; Lin, T.; Wu, H.; Tsai, K.C.; Li, K.; et al. Nucleostemin Modulates Outcomes of Hepatocellular Carcinoma via a Tumor Adaptive Mechanism to Genomic Stress. Mol. Cancer Res. 2020, 18, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Liu, X.; Kim, M.; Zhang, K.; Tsai, R.Y.L. Epigenome-wide analysis of aging effects on liver regeneration. BMC Biol. 2023, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, H.; Meng, L.; Lin, T.; Zhu, Q.; Tsai, R.Y. GNL3L inhibits activity of estrogen-related receptor-gamma by competing for coactivator binding. J. Cell Sci. 2007, 120, 2532–2543. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Hsu, J.K.; Zhu, Q.; Lin, T.; Tsai, R.Y. Nucleostemin inhibits TRF1 dimerization and shortens its dynamic association with the telomere. J. Cell Sci. 2011, 124, 3706–3714. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Meng, L.; Tsai, R.Y. p53 configures the G2/M arrest response of nucleostemin-deficient cells. Cell Death Discov. 2015, 1, e15060. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Meng, L.; Wu, L.J.; Pederson, T.; Tsai, R.Y. Nucleostemin and GNL3L exercise distinct functions in genome protection and ribosome synthesis, respectively. J. Cell Sci. 2014, 127, 2302–2312. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | Sense Primer | Anti-Sense Primer |
|---|---|---|
| MGMT | 5′-CAGACAGGTGTTATGGAAGCTGC-3′ | 5′-AGAAGCCATTCCTTCACGGCCAGT-3′ |
| TERT | 5′-ACGGTGTGCACCAACATCTAC-3′ | 5′-GCTTTCAGGATGGAGTAGCAG-3′ |
| EGFR | 5′-AGTCAGCAGTGACTTTCTCAGC-3′ | 5′-GGGCACAGATGATTTTGGTCAG-3′ |
| S6K1 | 5′-CAGCTCATACAAAAGCAGAACGG-3′ | 5′-GTAAAAGCAGGCAGTGTCTTCC-3′ |
| P14ART | 5′-GAGTGAGGGTTTTCGTGGTTCAC-3′ | 5′-CCCATCATCATGACCTGGTCTTC-3′ |
| P16INK4a | 5′-AGCATGGAGCCTTCGGCTGACTG-3′ | 5′-TGCCCATCATCATGACCTGGATC-3′ |
| TP53 | 5′-GCCAAAGAAGAAACCACTGGAT-3′ | 5′-GCCCTTCTGTCTTGAACATGAG-3′ |
| c-Myc | 5′-AGCGACTCTGAGGAGGAACAAG-3′ | 5′-TGCGTAGTTGTGCTGATGTGTG-3′ |
| STAT3 | 5′-AATCACGCCTTCTACAGACTGC-3′ | 5′-CATCCTGGAGATTCTCTACCAC-3′ |
| TFAP2A | 5′-CTCACATCACTAGTAGAGGGAG-3′ | 5′-AGCAGGTCGGTGAACTCTTTG-3′ |
| CTCF | 5′-AGTGTTCCATGTGCGATTACGC-3′ | 5′-TTCAGCTTGTATGTGTCCCTGC-3′ |
| GABPA | 5′-ACCACACTCAACATTTCGGGG-3′ | 5′-AGCAGATTGCACTGATGCTGG-3′ |
| HMGN1 | 5′-GGCAGCAGCGAAGGATAAATC-3′ | 5′-TTCATCAGAGGCTGGACTCTC-3′ |
| GAPDH | 5′-GTGAAGGTCGGAGTCAAC-3′ | 5′-TTCTCAGCCTTGACGGTG-3′ |
| TMZ | IRI | BCNU | MPA | OXP | Taxol | |
|---|---|---|---|---|---|---|
| U87 | 600 μM | 20 μM | 200 μM | 120 μM | 15 μM | 50 nM |
| U251 | 400 μM | 10 μM | 50 μM | 120 μM | 10 μM | 0.3 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Wang, J.; Wu, L.J.; Trinh, B.; Tsai, R.Y.L. IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells. Int. J. Mol. Sci. 2024, 25, 5992. https://doi.org/10.3390/ijms25115992
Liu X, Wang J, Wu LJ, Trinh B, Tsai RYL. IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells. International Journal of Molecular Sciences. 2024; 25(11):5992. https://doi.org/10.3390/ijms25115992
Chicago/Turabian StyleLiu, Xiaoqin, Junying Wang, Laura J. Wu, Britni Trinh, and Robert Y. L. Tsai. 2024. "IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells" International Journal of Molecular Sciences 25, no. 11: 5992. https://doi.org/10.3390/ijms25115992
APA StyleLiu, X., Wang, J., Wu, L. J., Trinh, B., & Tsai, R. Y. L. (2024). IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells. International Journal of Molecular Sciences, 25(11), 5992. https://doi.org/10.3390/ijms25115992