
Specific Mutations Reverse Regulatory Effects of Adenosine Phosphates and Increase Their Binding Stoichiometry in CBS Domain-Containing Pyrophosphatase


Abstract
1. Introduction
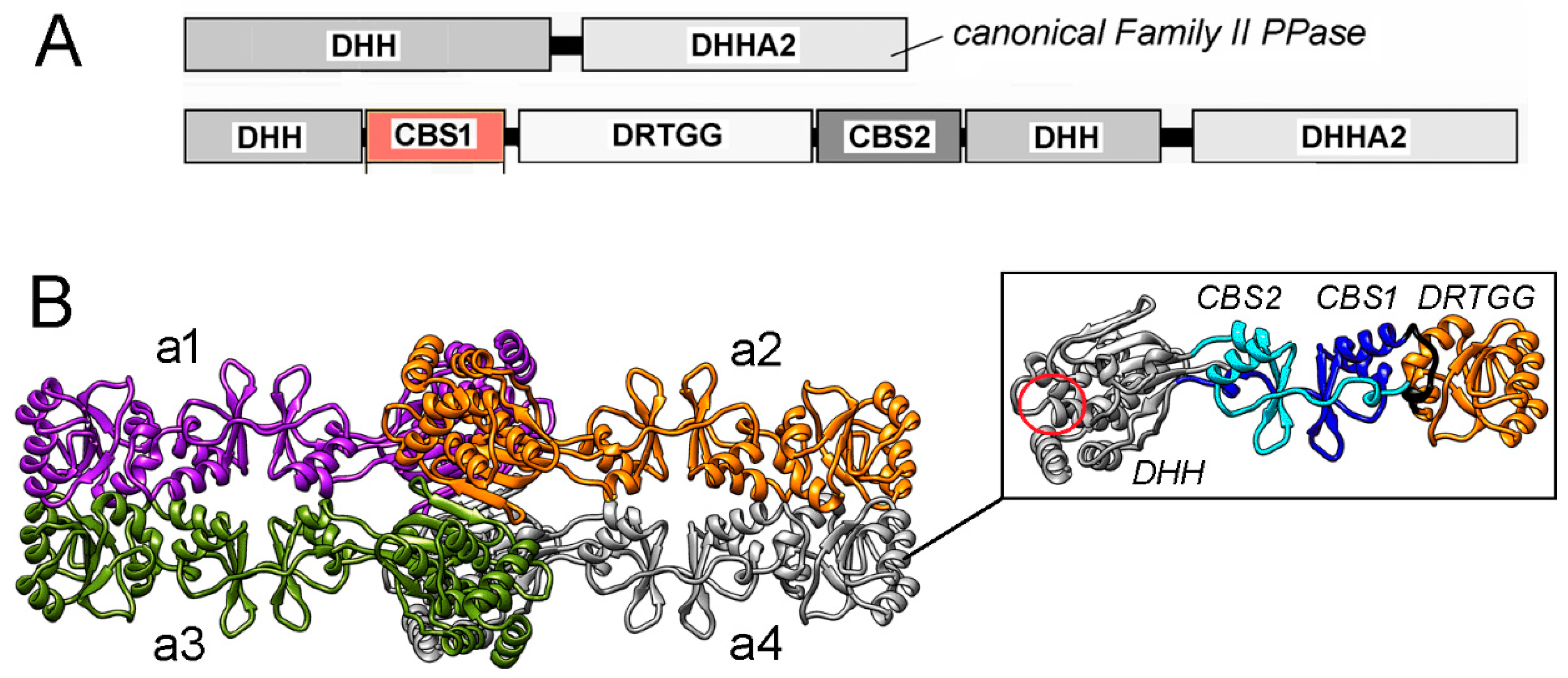
2. Results
2.1. Selection of Residues for Substitution
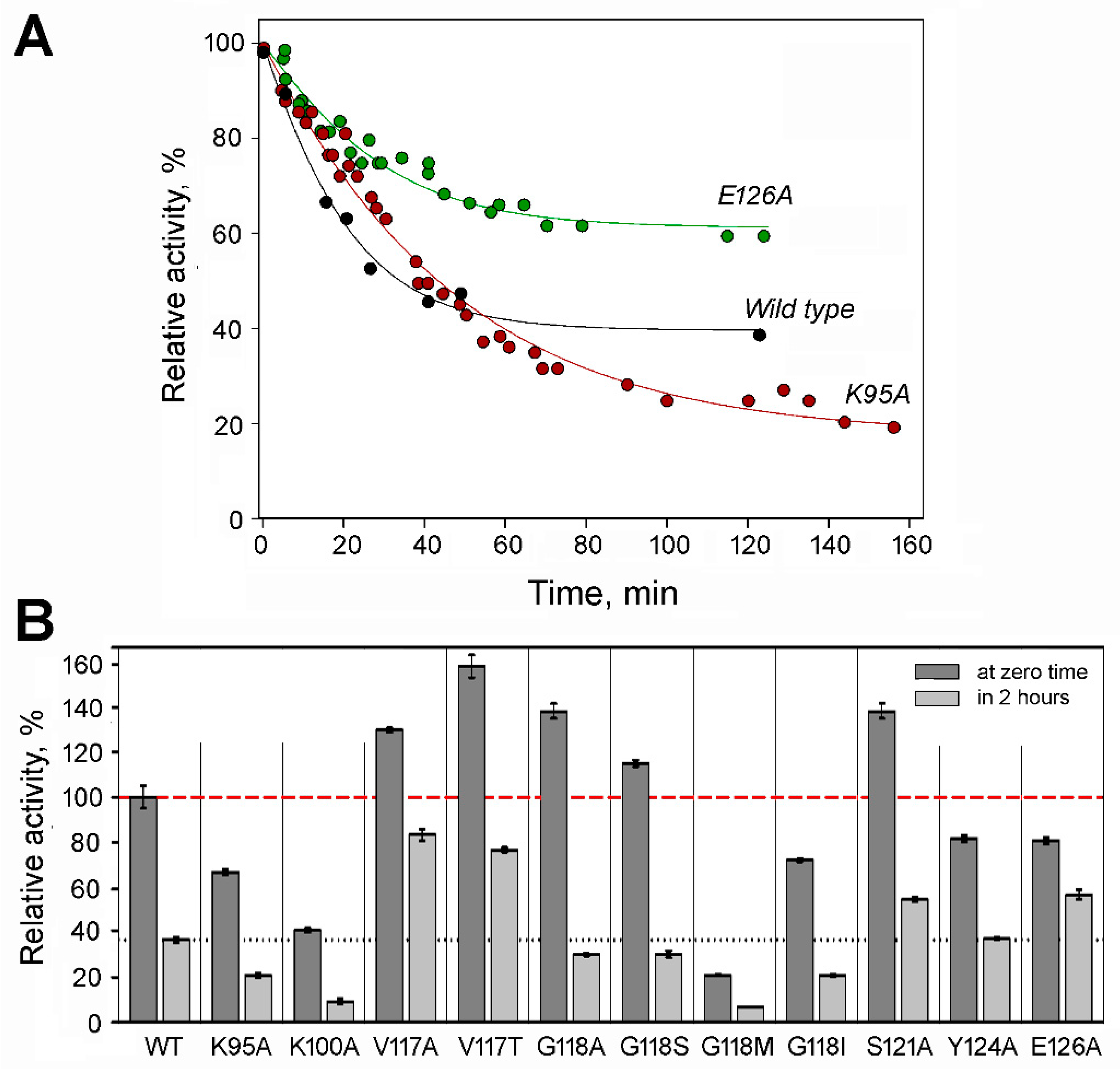
2.2. Effects of Substitutions on Tetramer Activity and Stability
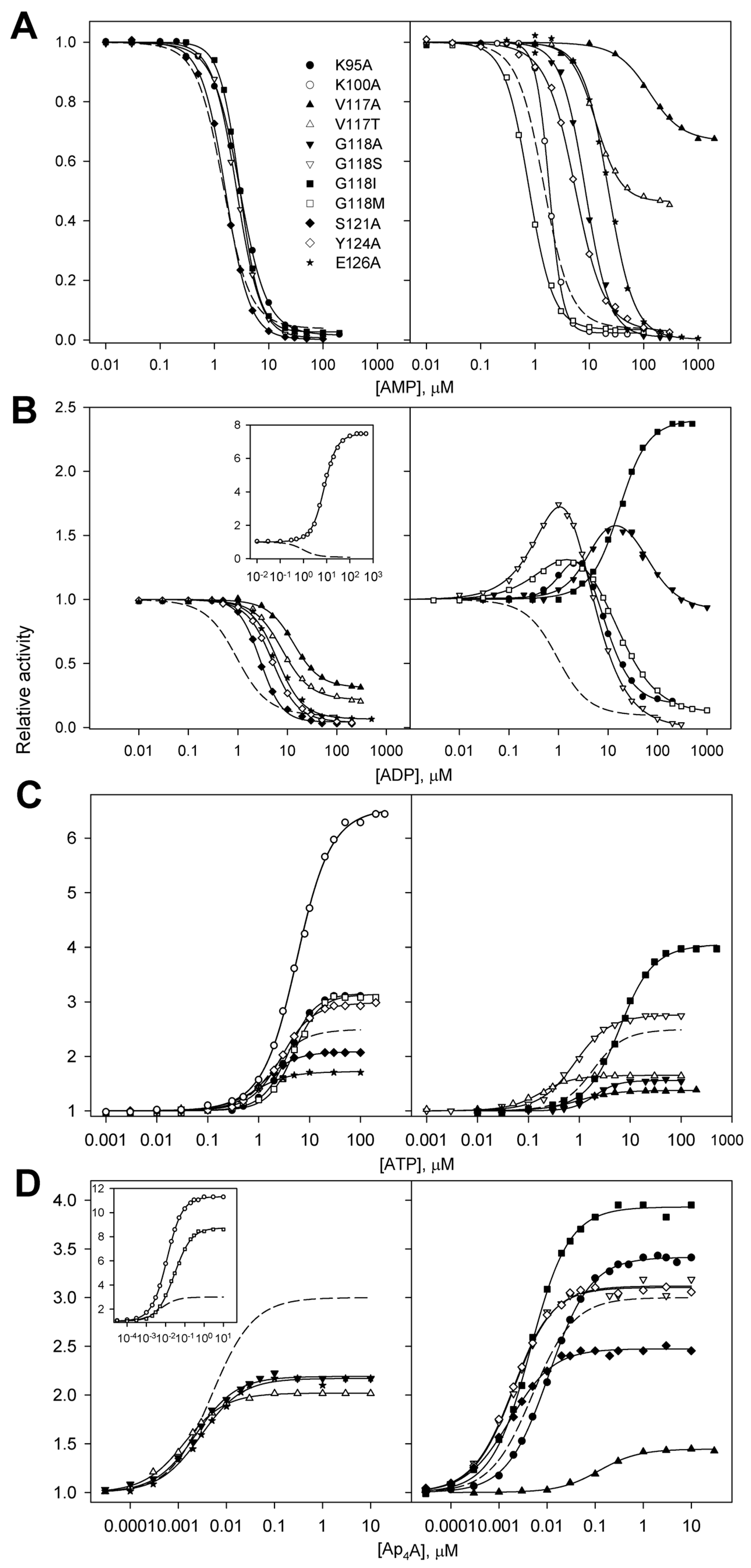
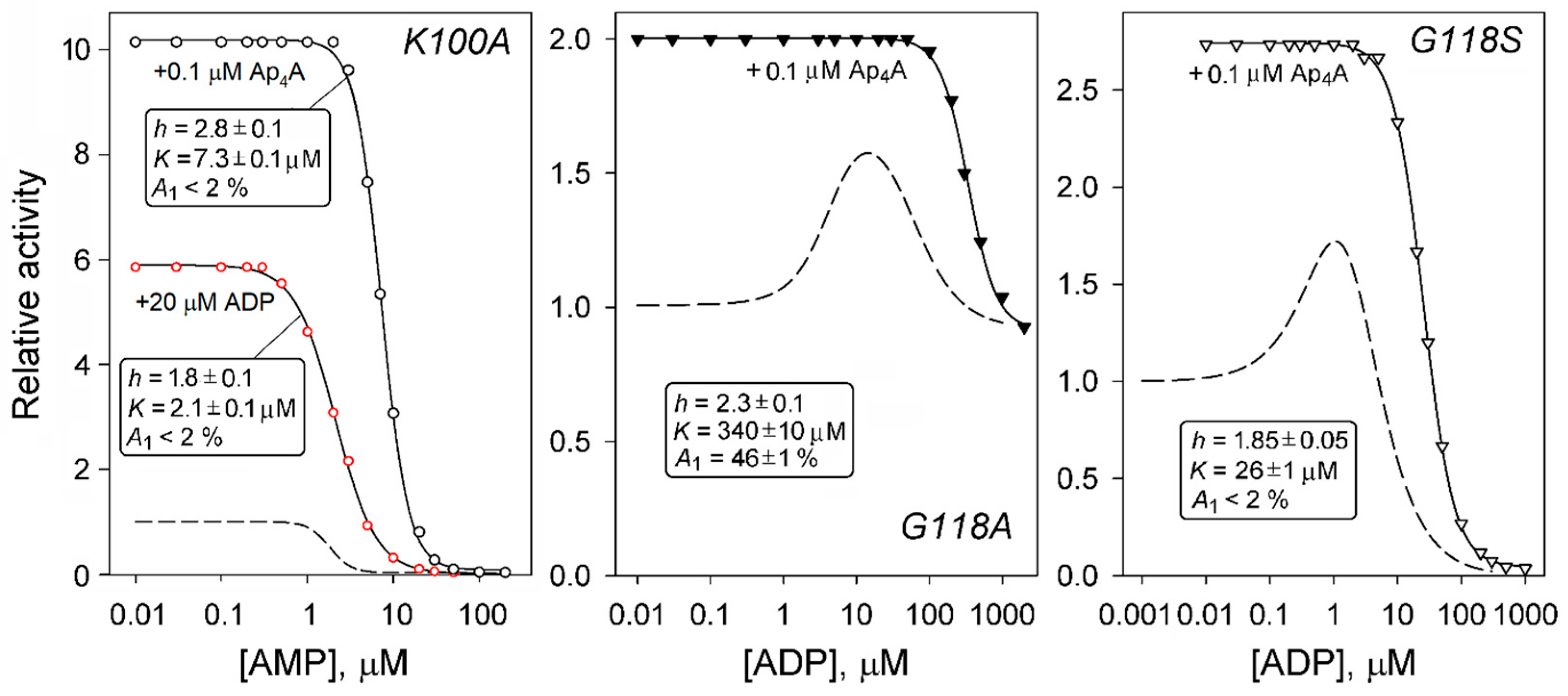
2.3. Regulation of dhPPase Variants by Adenosine Phosphates
2.4. AMP/ADP and ADP/Ap4A Competition in Variants with Unusual ADP Effects
2.5. Measurements of Adenosine Phosphate Binding by Isothermal Titration Calorimetry (ITC)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Enzyme Activity Assay
4.3. Isothermal Titration Calorimetry (ITC)
4.4. Structure Modeling and Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bateman, A. The structure of a domain common to Archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Labesse, G.; Alexandre, T.; Vaupre, L.; Salard-Arnaud, I.; Him, J.L.; Raynal, B.; Bron, P.; Munier-Lehmann, H. MgATP regulates allostery and fiber formation in IMPDHs. Structure 2013, 21, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Chen, Z.P.; Van Denderen, B.J.; Morton, C.J.; Parker, M.W.; Witters, L.A.; Stapleton, D.; Kemp, B.E. Intrasteric control of AMPK via the c1 subunit AMP allosteric regulatory site. Protein Sci. 2004, 13, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Justel, D.; Marcos-Alcalde, I.; Abascal, F.; Vidana, N.; Gomez-Puertas, P.; Jimenez, A.; Revuelta, J.L.; Buey, R.M. Diversity of mechanisms to control bacterial GTP homeostasis by the mutually exclusive binding of adenine and guanine nucleotides to IMP dehydrogenase. Protein Sci. 2022, 31, e4314. [Google Scholar] [CrossRef] [PubMed]
- Jämsen, J.; Tuominen, H.; Salminen, A.; Belogurov, G.A.; Magretova, N.N.; Baykov, A.A.; Lahti, R. A CBS domain-containing pyrophosphatase of Moorella thermoacetica is regulated by adenine nucleotides. Biochem. J. 2007, 408, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kery, V.; Poneleit, L.; Kraus, J. Trypsin cleavage of human cystathionine β-synthase into an evolutionarily conserved active core: Structural and functional consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Anashkin, V.A.; Salminen, A.; Lahti, R. Inorganic pyrophosphatases of Family II—Two decades after their discovery. FEBS Lett. 2017, 591, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Anashkin, V.A.; Lahti, M.; Tuominen, H.K.; Lahti, R.; Baykov, A.A. Cystathionine β-synthase (CBS) domains confer multiple forms of Mg2+-dependent cooperativity to family II pyrophosphatases. J. Biol. Chem. 2014, 289, 22865–22876. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Salminen, A.; Tuominen, H.K.; Orlov, V.N.; Lahti, R.; Baykov, A.A. Cystathionine β-synthase (CBS) domain-containing pyrophosphatase as a target for diadenosine polyphosphates in bacteria. J. Biol. Chem. 2015, 290, 27594–27603. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Salminen, A.; Orlov, V.N.; Lahti, R.; Baykov, A.A. The tetrameric structure of nucleotide-regulated pyrophosphatase and its modulation by deletion mutagenesis and ligand binding. Arch. Biochem. Biophys. 2022, 692, 108537. [Google Scholar] [CrossRef]
- Zamakhov, I.M.; Anashkin, V.A.; Moiseenko, A.V.; Orlov, V.N.; Sokolova, O.S.; Baykov, A.A. The structure and nucleotide-binding characteristics of regulated CBS domain-containing pyrophosphatase without one catalytic domain. Int. J. Mol. Sci. 2023, 24, 17160. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, H.; Salminen, A.; Oksanen, E.; Jämsen, J.; Heikkilä, O.; Lehtio, L.; Magretova, N.N.; Goldman, A.; Baykov, A.A.; Lahti, R. Crystal structures of the CBS and DRTGG domains of the regulatory region of Clostridium perfringens pyrophosphatase complexed with the inhibitor, AMP, and activator, diadenosine tetraphosphate. J. Mol. Biol. 2010, 398, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Orlov, V.N.; Lahti, R.; Baykov, A.A. An arginine residue involved in allosteric regulation of cystathionine β-synthase (CBS) domain-containing pyrophosphatase. Arch. Biochem. Biophys. 2019, 662, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Anu, S.; Osipova, E.; Kurilova, S.A.; Deltsov, I.D.; Reijo, L.; Baykov, A.A. Residue network involved in the allosteric regulation of cystathionine β-synthase domain-containing pyrophosphatase by adenine nucleotides. ACS Omega 2019, 4, 15549–15559. [Google Scholar] [CrossRef] [PubMed]
- Ereno-Orbea, J.; Oyenarte, I.; Martìnez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Day, P.; Sharff, A.; Parra, L.; Cleasby, A.; Williams, M.; Hörer, S.; Nar, H.; Redemann, N.; Tickle, I.; Yon, J. Structure of a CBS domain pair from the regulatory γ1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Purich, D.L. Enzyme Kinetics; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 691–709. [Google Scholar]
- Forsén, S.; Linse, S. Cooperativity: Over the Hill. Trends Biochem. Sci. 1995, 20, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Aksenova, V.A.; Vorobyeva, N.N.; Baykov, A.A. Roles of nucleotide substructures in the regulation of cystathionine β-synthase domain-containing pyrophosphatase. Biochim. Biophys. Acta—Gen. Subj. 2019, 1863, 1263–1269. [Google Scholar] [CrossRef]
- Jämsen, J.; Tuominen, H.; Baykov, A.A.; Lahti, R. Mutational analysis of residues in the regulatory CBS domains of Moorella thermoacetica pyrophosphatase corresponding to disease-related residues of human proteins. Biochem. J. 2011, 435, 497–504. [Google Scholar] [CrossRef]
- Barnes, B.R.; Marklund, S.; Steiler, T.L.; Walter, M.; Hjälm, G.; Amarger, V.; Mahlapuu, M.; Leng, Y.; Johansson, C.; Galuska, D.; et al. The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 2004, 279, 38441–38447. [Google Scholar] [CrossRef]
- Gollob, M.H.; Green, M.S.; Tang, A.S.; Gollob, T.; Karibe, A.; Ali Hassan, A.S.; Ahmad, F.; Lozado, R.; Shah, G.; Fananapazir, L.; et al. Identification of a gene responsible for familial Wolff–Parkinson–White syndrome. N. Engl. J. Med. 2001, 344, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Costford, S.; Kavaslar, N.; Ahituv, N.; Chaudhry, S.; Schackwitz, W.; Dent, R.; Pennacchio, L.; McPherson, R.; Harper, M. Gain-of-function R225W mutation in human AMPKγ3 causing increased glycogen and decreased triglyceride in skeletal muscle. PLoS ONE 2007, 2, e903. [Google Scholar] [CrossRef] [PubMed]
- Kluijtmans, L.A.; Boers, G.H.; Stevens, E.M.; Renier, W.O.; Kraus, J.P.; Trijbels, F.J.; Van Den Heuvel, L.P.; Blom, H.J. Defective cystathionineβ-synthase regulation by S-adenosylmethionine in a partially pyridoxine responsive homocystinuria patient. J. Clin. Investig. 1996, 98, 285–289. [Google Scholar] [CrossRef]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.; Leone, P.; Jing, C.; Walker, P.; Haire, L.; Eccleston, J.; Davis, C.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Townley, R.; Shapiro, L. Structural insight into AMPK regulation: ADP comes into play. Structure 2007, 15, 1285–1295. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef]
- Gomez-García, I.; Oyenarte, I.; Martínez-Cruz, L. The crystal structure of protein MJ1225 from Methanocaldococcus jannaschii shows strong conservation of key structural features seen in the eukaryal γ-AMPK. J. Mol. Biol. 2010, 399, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J.; Zhang, Y.Y.; Neumann, D.; Schlattner, U.; Wang, Z.-X.; Wu, J.-W. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 2012, 19, 716–718. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martίnez-Cruz, L.A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proc. Natl Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef]
- Meyer, S.; Savaresi, S.; Forster, I.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, J.K. Biological Role of Inorganic Pyrophosphate; Kluwer Academic Publishers: London, UK, 2001. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Avaeva, S.M. A simple and sensitive apparatus for continuous monitoring of orthophosphate in the presence of acid-labile compounds. Anal. Biochem. 1981, 116, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]

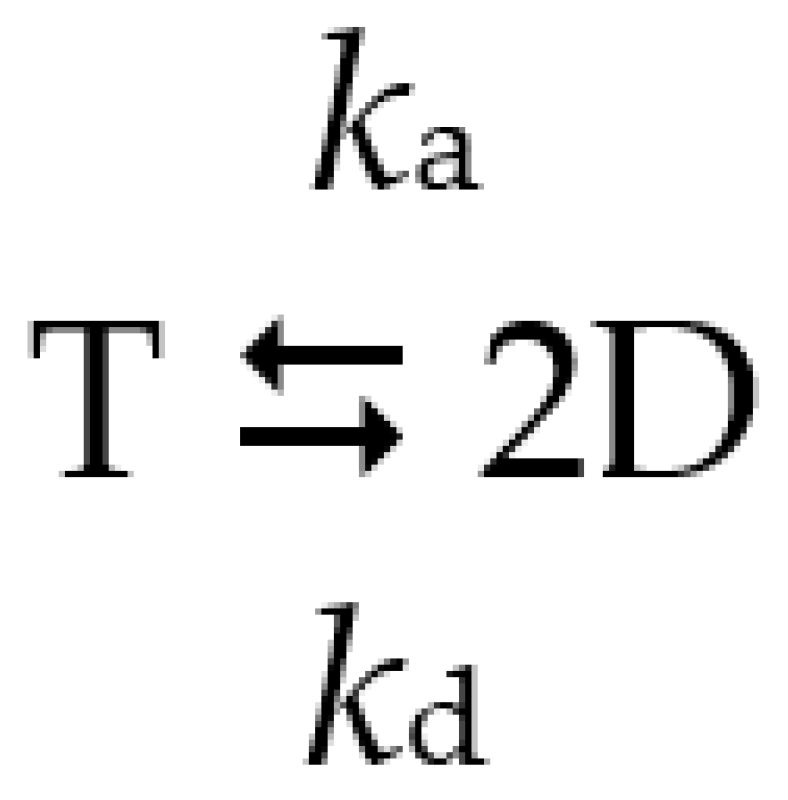


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Variant | AT, % | kd, h−1 | ka, μM−1·h−1 | Kd, μM |
|---|---|---|---|---|
| WT | 100 ± 5 | 1.6 ± 0.1 (1.8 ± 0.1) a | 2.1 ± 0.5 (2.4 ± 0.4) a | 0.8 ± 0.2 (0.7 ± 0.2) a |
| K95A | 66 ± 2 | 1.00 ± 0.04 | 0.3 ± 0.1 | 3.2 ± 0.8 |
| K100A | 42 ± 1 | 1.1 ± 0.1 | 0.19 ± 0.04 | 5.9 ± 1.6 |
| V117A | 113 ± 1 | 0.44 ± 0.02 | 1.5 ± 0.2 | 0.3 ± 0.1 |
| V117T | 141 ± 5 | 0.6 ± 0.1 | 0.6 ± 0.3 | 1.0 ± 0.6 |
| G118A | 120 ± 3 | 1.06 ± 0.04 | 0.15 ± 0.04 | 6.8 ± 0.6 |
| G118S | 117 ± 2 | 0.85 ± 0.04 | 0.14 ± 0.06 | 6.0 ± 2.7 |
| G118M | 21 ± 1 | 0.70 ± 0.04 | 0.18 ± 0.10 | 4 ± 2 |
| G118I | 73 ± 1 | 0.75 ± 0.03 | 0.12 ± 0.05 | 6.2 ± 2.9 |
| S121A | 120 ± 3 | 1.5 ± 0.1 | 0.5 ± 0.1 | 3.3 ± 0.9 |
| Y124A | 83 ± 2 | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.2 |
| E126A | 82 ± 2 | 0.7 ± 0.1 | 3.4 ± 0.5 | 0.21 ± 0.05 |
| Enzyme Variant | Adenosine Phosphate and Parameter Value a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AMP | ADP | ATP | Ap4A | |||||||||
| A1, % | K, µM | h | A1, % | K, µM | h | A1, % | K, µM | h | A1, % | K, nM | h | |
| WT b | 3.7 ± 0.1 | 1.4 ± 0.1 | 1.7 ± 0.1 | 9 ± 1 | 0.95 ± 0.05 | 1.34 ± 0.03 | 250 ± 20 | 1.9 ± 0.2 | 1.45 ± 0.13 | 300 ± 10 | 4.9 ± 0.2 | 1.00 ± 0.02 |
| K95A | 1.4 ± 0.2 | 2.90 ± 0.04 | 1.7 ± 0.1 | N.A. c | N.A. | N.A. | 317 ± 5 | 3.9 ± 0.1 | 1.63 ± 0.06 | 341 ± 6 | 10.9 ± 0.3 | 1.00 ± 0.02 |
| K100A | 2.3 ± 0.4 | 1.82 ± 0.02 | 3.5 ± 0.1 | 750 ± 30 | 7.8 ± 0.2 | 1.55 ± 0.05 | 650 ± 20 | 5.6 ± 0.1 | 1.26 ± 0.04 | 1130 ± 20 | 11.5 ± 0.1 | 1.07 ± 0.01 |
| V117A | 66 ± 1 | 124 ± 5 | 1.4 ± 0.1 | 31 ± 1 | 13.4 ± 0.5 | 1.60 ± 0.08 | 138 ± 1 | 0.95 ± 0.05 | 1.22 ± 0.07 | 145 ± 2 | 120 ± 10 | 1.06 ± 0.09 |
| V117T | 46 ± 1 | 13.2 ± 0.4 | 1.9 ± 0.1 | 21 ± 1 | 7.9 ± 0.3 | 1.48 ± 0.07 | 165 ± 4 | 0.17 ± 0.02 | 1.34 ± 0.12 | 202 ± 2 | 1.30 ± 0.05 | 1.02 ± 0.04 |
| G118A | 0.8 ± 0.7 | 8.4 ± 0.2 | 1.9 ± 0.1 | N.A. | N.A. | N.A. | 156 ± 2 | 2.00 ± 0.07 | 1.92 ± 0.12 | 219 ± 4 | 2.3 ± 0.2 | 1.02 ± 0.06 |
| G118S | 0.5 ± 0.6 | 2.55 ± 0.04 | 2.0 ± 0.1 | N.A. | N.A. | N.A. | 277 ± 5 | 0.82 ± 0.03 | 1.25 ± 0.05 | 312 ± 11 | 1.9 ± 0.2 | 1.03 ± 0.08 |
| G118M | 3.4 ± 0.6 | 0.76 ± 0.02 | 1.9 ± 0.1 | N.A. | N.A. | N.A. | 314 ± 8 | 4.9 ± 0.2 | 1.70 ± 0.12 | 870 ± 30 | 28 ± 1 | 0.99 ± 0.02 |
| G118I | 1.6 ± 1.0 | 2.90 ± 0.03 | 2.2 ± 0.1 | 240 ± 3 | 17 ± 1 | 1.5 ± 0.1 | 410 ± 12 | 6.2 ± 0.2 | 1.30 ± 0.06 | 393 ± 9 | 4.3 ± 0.2 | 1.05 ± 0.04 |
| S121A | 1 ± 4 | 1.60 ± 0.02 | 1.9 ± 0.1 | 4.2 ± 0.9 | 2.8 ± 0.1 | 2.0 ± 0.10 | 209 ± 2 | 1.27 ± 0.03 | 1.41 ± 0.05 | 247 ± 4 | 1.6 ± 0.1 | 0.97 ± 0.05 |
| Y124A | 2.6 ± 0.6 | 5.4 ± 0.1 | 1.6 ± 0.1 | 3.8 ± 0.6 | 5.0 ± 0.1 | 1.63 ± 0.04 | 299 ± 4 | 2.81 ± 0.06 | 1.40 ± 0.04 | 311 ± 7 | 1.8 ± 0.1 | 1.04 ± 0.05 |
| E126A | 0.2 ± 0.9 | 22.5 ± 0.5 | 1.8 ± 0.1 | 6.5 ± 1.6 | 6.2 ± 0.3 | 1.6 ± 0.1 | 172 ± 2 | 0.80 ± 0.06 | 1.25 ± 0.10 | 217 ± 4 | 2.9 ± 0.2 | 1.02 ± 0.07 |
| Enzyme Variant | A1, % | A2, % | K1, μM | K2, μM | h1 | h2 |
|---|---|---|---|---|---|---|
| K95A | 170 ± 30 | 18 ± 2 | 1.7 ± 0.6 | 7 ± 2 | 1.8 ± 0.3 | 1.4 ± 0.2 |
| G118A | 180 ± 20 | 91 ± 5 | 4.8 ± 1.5 | 50 ± 20 | 1.5 ± 0.2 | 1.3 ± 0.2 |
| G118S | 270 ± 30 | 0.8 ± 1.1 | 0.8 ± 0.2 | 3.0 ± 0.6 | 1.1 ± 0.1 | 1.1 ± 0.1 |
| G118M | 174 ± 19 | 10 ± 1 | 1.0 ± 0.5 | 10 ± 2 | 0.9 ± 0.1 | 0.9 ± 0.1 |
| Enzyme Variant | Adenosine Phosphate | ΔH, kcal/mol | n | KN, µM | −TΔS, kcal/mol |
|---|---|---|---|---|---|
| WT | AMP | −5.6 ± 0.5 | 0.79 ± 0.05 | 0.8 ± 0.3 | −2.7 ± 0.9 |
| K95A | AMP | −2.6 ± 0.2 | 2.41 ± 0.04 | 4.6 ± 0.7 | −4.7 ± 0.4 |
| K100A | AMP | −4.3 ± 0.1 | 1.85 ± 0.04 | 1.3 ± 0.2 | −3.7 ± 0.3 |
| V117A | AMP | −4.1 ± 0.6 | 0.94 ± 0.12 | 4.4 ± 0.8 | −3.2 ± 0.8 |
| V117T | AMP | −5.5 ± 0.2 | 0.94 ± 0.03 | 1.7 ± 0.2 | −2.4 ± 0.3 |
| G118A | AMP | −2.2 ± 0.1 | 1.96 ± 0.09 | 4.2 ± 0.6 | −5.2 ± 0.2 |
| G118S | AMP | −3.6 ± 0.2 | 1.16 ± 0.06 | 2.8 ± 0.4 | −4.0 ± 0.3 |
| G118M | AMP | −4.0 ± 0.1 | 0.98 ± 0.02 | 0.52 ± 0.07 | −4.5 ± 0.2 |
| G118I | AMP | 4.6 ± 0.2 | 1.07 ± 0.03 | 1.6 ± 0.2 | −3.3 ± 0.3 |
| S121A | AMP | −5.1 ± 0.2 | 1.07 ± 0.02 | 1.1 ± 0.10 | −3.1 ± 0.3 |
| Y124A | AMP | −3.0 ± 0.2 | 1.10 ± 0.04 | 2.5 ± 0.2 | −4.7 ± 0.2 |
| E126A | AMP | −3.8 ± 0.2 | 1.05 ± 0.04 | 1.9 ± 0.4 | −4.0 ± 0.4 |
| WT | ADP | −5.9 ± 0.3 | 0.90 ± 0.04 | 1.0 ± 0.2 | −2.3 ± 0.5 |
| K95A | ADP | −3.6 ± 0.2 | 2.1 ± 0.10 | 9.3 ± 1.7 | −3.2 ± 0.4 |
| K100A | ADP | −1.6 ± 0.2 | 2.1 ± 0.2 | 6 ± 2 | −5.5 ± 0.5 |
| G118A | ADP | −3.3 ± 0.3 | 2.1 ± 0.1 | 5 ± 1 | −4.0 ± 0.5 |
| G118S | ADP | −5.8 ± 0.2 | 1.18 ± 0.03 | 1.1 ± 0.2 | −2.3 ± 0.4 |
| E126A | ADP | −5.9 ± 0.2 | 0.97 ± 0.02 | 0.6 ± 0.1 | −2.6 ± 0.3 |
| WT | ATP | −6.8 ± 0.2 | 0.88 ± 0.02 | 2.1 ± 0.3 | −0.9 ± 0.3 |
| K95A | ATP | −4.3 ± 0.7 | 2.4 ± 0.3 | 6 ± 2 | −2.8 ± 1.0 |
| K100A | ATP | −0.41 ± 0.03 | 1.9 ± 0.1 | 0.4 ± 0.2 | −8.3 ± 0.5 |
| G118A | ATP | −1.3 ± 0.4 | 2.0 ± 0.3 | 3 ± 4 | −6.3 ± 1.7 |
| G118S | ATP | −1.0 ± 0.2 | 1.1 ± 0.1 | 0.5 ± 0.5 | −8 ± 1 |
| E126A | ATP | −4.1 ± 0.6 | 1.0 ± 0.1 | 4.3 ± 1.2 | −3.2 ± 0.9 |
| WT | Ap4A | −10.3 ± 0.3 | 0.41 ± 0.01 | N.D. b | N.A. c |
| K95A | Ap4A | −13.7 ± 0.6 | 0.45 ± 0.01 | N.D. | N.A. |
| K100A | Ap4A | −11.4 ± 0.4 | 0.52 ± 0.01 | N.D. | N.A. |
| V117A | Ap4A | −9.0 ± 0.3 | 0.41 ± 0.01 | N.D. | N.A. |
| V117T | Ap4A | −9.1 ± 0.4 | 0.41 ± 0.01 | N.D. | N.A. |
| G118A | Ap4A | −10.0 ± 0.2 | 0.47 ± 0.01 | N.D. | N.A. |
| G118S | Ap4A | −8.0 ± 0.2 | 0.40 ± 0.01 | N.D. | N.A. |
| G118M | Ap4A | −9.2 ± 0.4 | 0.45 ± 0.01 | N.D. | N.A. |
| G118I | Ap4A | −10.0 ± 0.4 | 0.44 ± 0.01 | N.D. | N.A. |
| S121A | Ap4A | −14.3 ± 0.3 | 0.42 ± 0.01 | N.D. | N.A. |
| Y124A | Ap4A | −10.0 ± 0.4 | 0.41 ± 0.01 | N.D. | N.A. |
| E126A | Ap4A | −10.7 ± 0.2 | 0.40 ± 0.01 | N.D. | N.A. |
| Residue | Direction of ADP Effect | Effect Size | Binding Stoichiometry | Binding Affinity | Binding Cooperativity | Tetramer Stability |
|---|---|---|---|---|---|---|
| Lys95 | ++ | +++ | − − | |||
| Lys100 | +++ | +++ | +++ | +++ | − − | |
| Val117 | + | − − − | ++ | + | ||
| Gly118 | +++ | ++ | + | + | +++ | − − |
| Ser121 | − | |||||
| Tyr124 | + | |||||
| Glu126 | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anashkin, V.A.; Kirillova, E.A.; Orlov, V.N.; Baykov, A.A. Specific Mutations Reverse Regulatory Effects of Adenosine Phosphates and Increase Their Binding Stoichiometry in CBS Domain-Containing Pyrophosphatase. Int. J. Mol. Sci. 2024, 25, 5768. https://doi.org/10.3390/ijms25115768
Anashkin VA, Kirillova EA, Orlov VN, Baykov AA. Specific Mutations Reverse Regulatory Effects of Adenosine Phosphates and Increase Their Binding Stoichiometry in CBS Domain-Containing Pyrophosphatase. International Journal of Molecular Sciences. 2024; 25(11):5768. https://doi.org/10.3390/ijms25115768
Chicago/Turabian StyleAnashkin, Viktor A., Elena A. Kirillova, Victor N. Orlov, and Alexander A. Baykov. 2024. "Specific Mutations Reverse Regulatory Effects of Adenosine Phosphates and Increase Their Binding Stoichiometry in CBS Domain-Containing Pyrophosphatase" International Journal of Molecular Sciences 25, no. 11: 5768. https://doi.org/10.3390/ijms25115768
APA StyleAnashkin, V. A., Kirillova, E. A., Orlov, V. N., & Baykov, A. A. (2024). Specific Mutations Reverse Regulatory Effects of Adenosine Phosphates and Increase Their Binding Stoichiometry in CBS Domain-Containing Pyrophosphatase. International Journal of Molecular Sciences, 25(11), 5768. https://doi.org/10.3390/ijms25115768