
Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
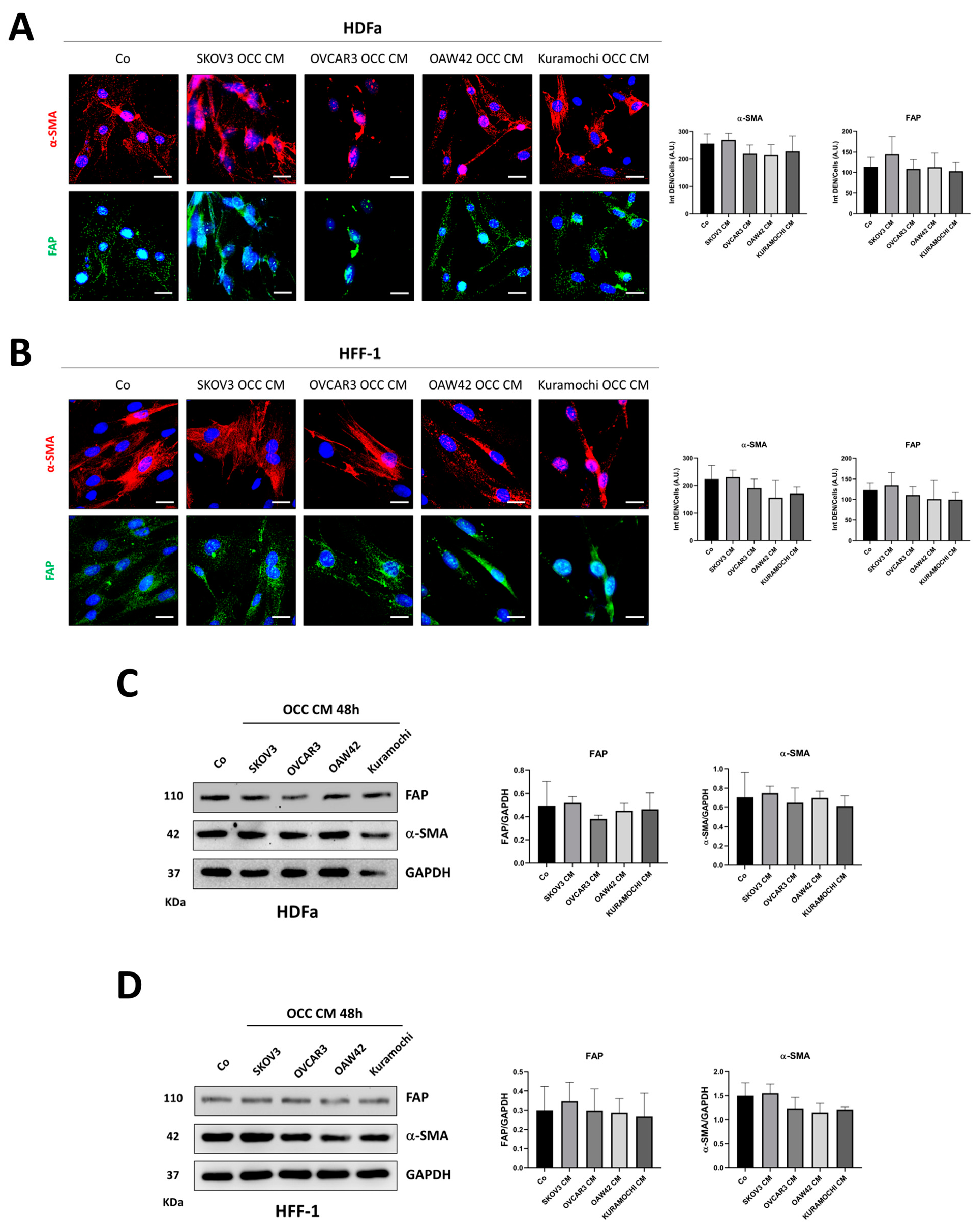
2.1. The Secretion from Different Ovarian Cancer Cells Promotes the Phenoconversion of Normal Skin Fibroblasts (NFs) into Cancer-Associated Fibroblasts (CAFs)
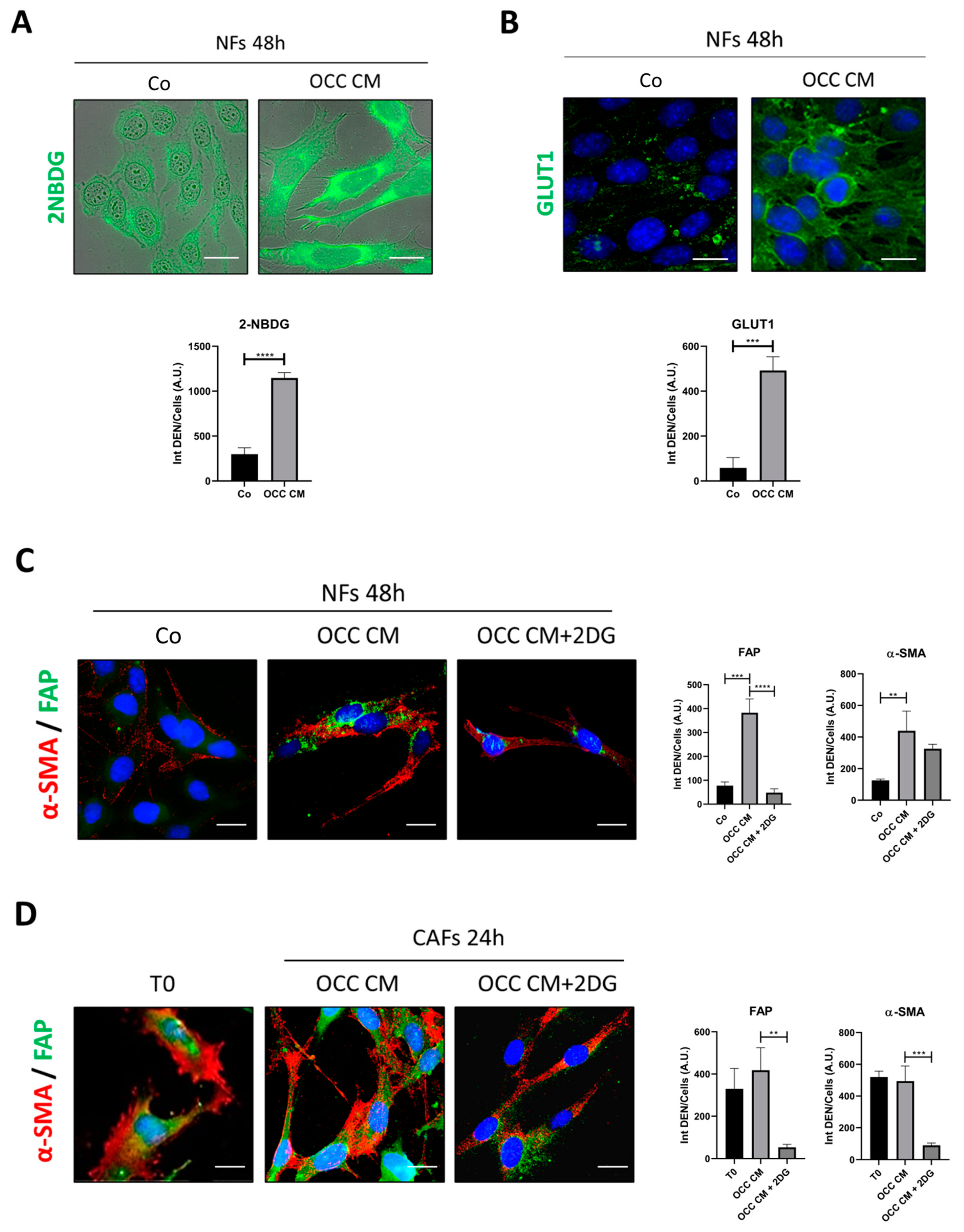
2.2. CAFs Phenoconversion Requires the Glycolytic Shift and Can Be Reversed through Glycolysis Inhibition
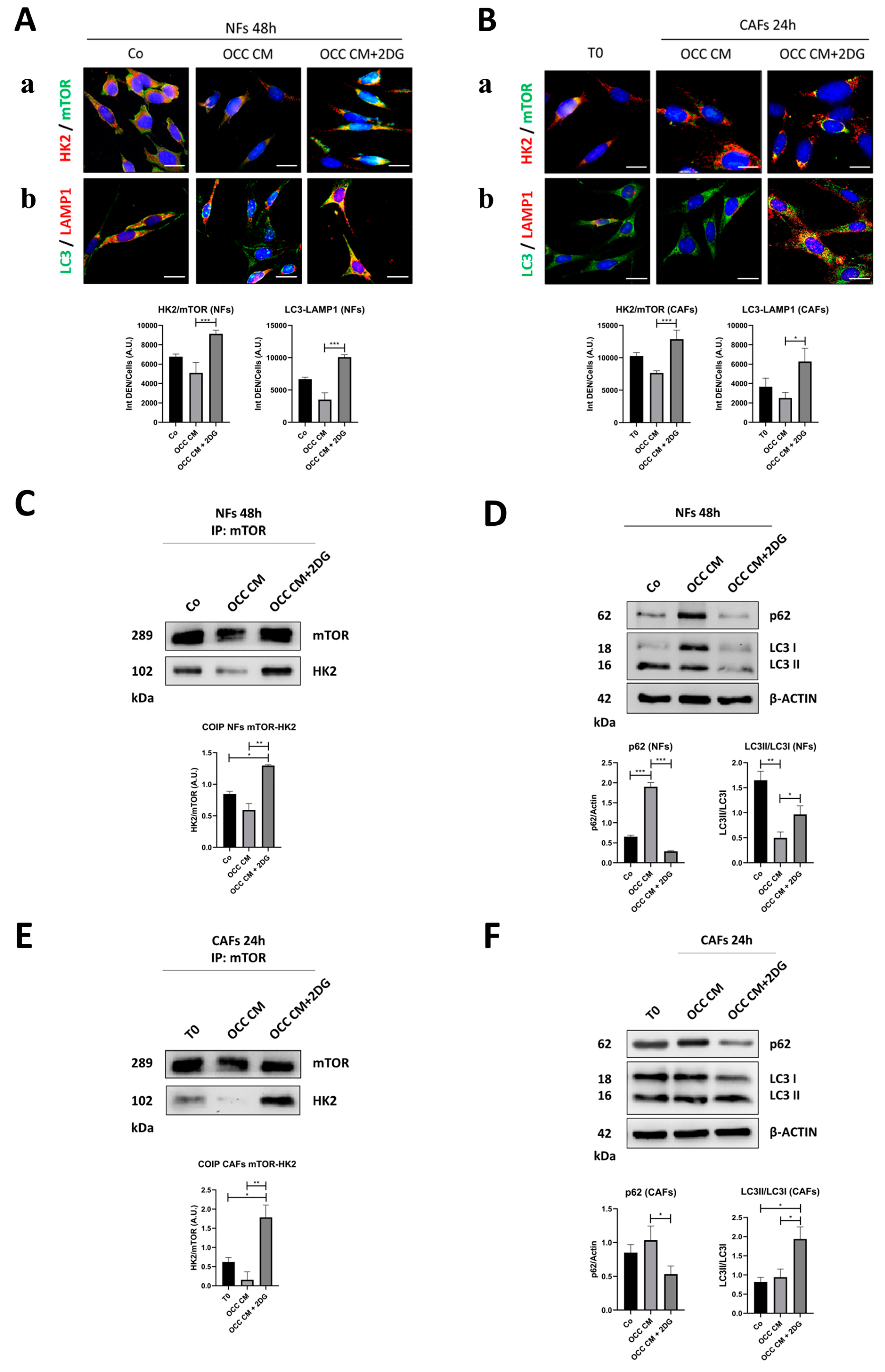
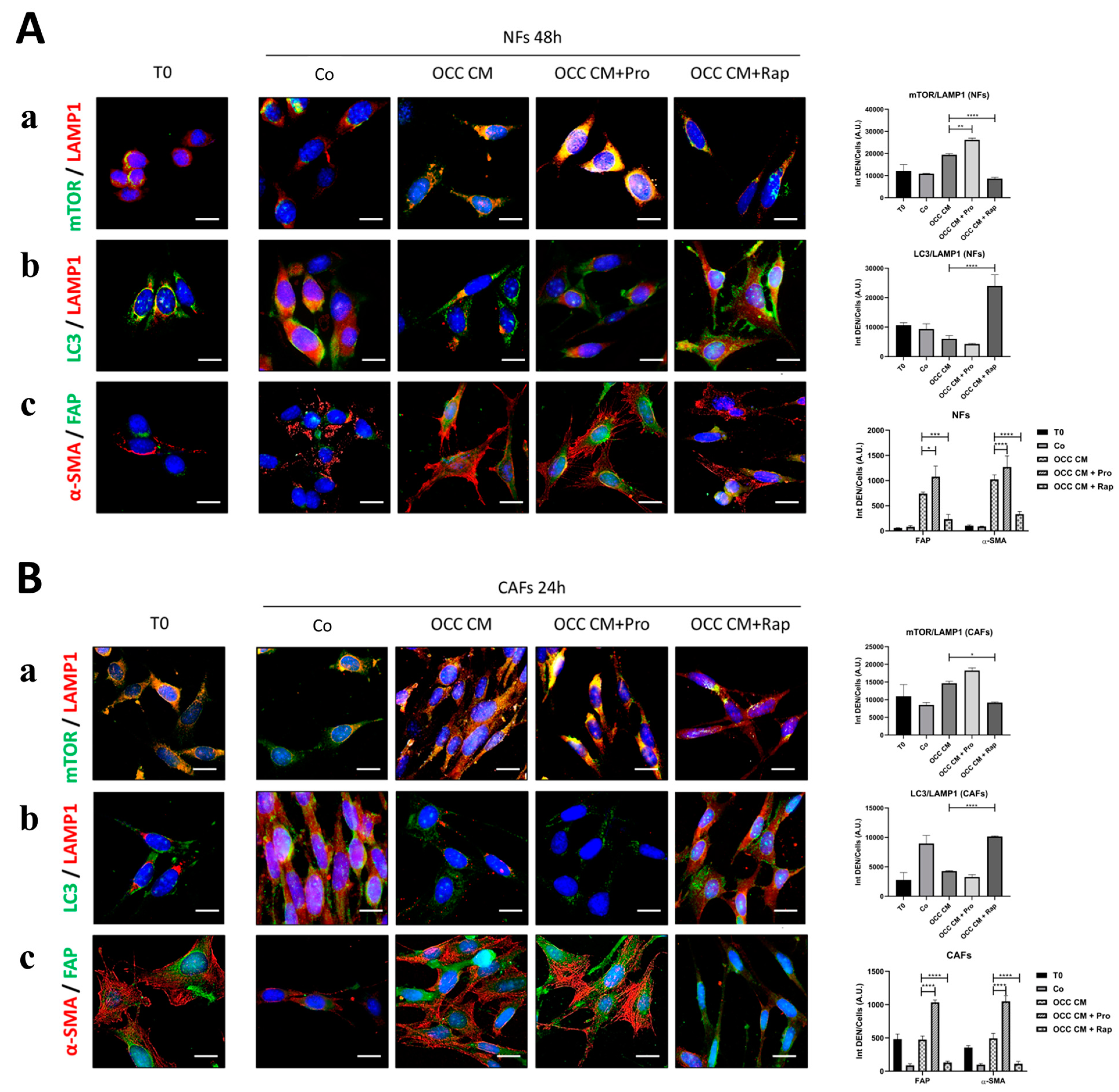
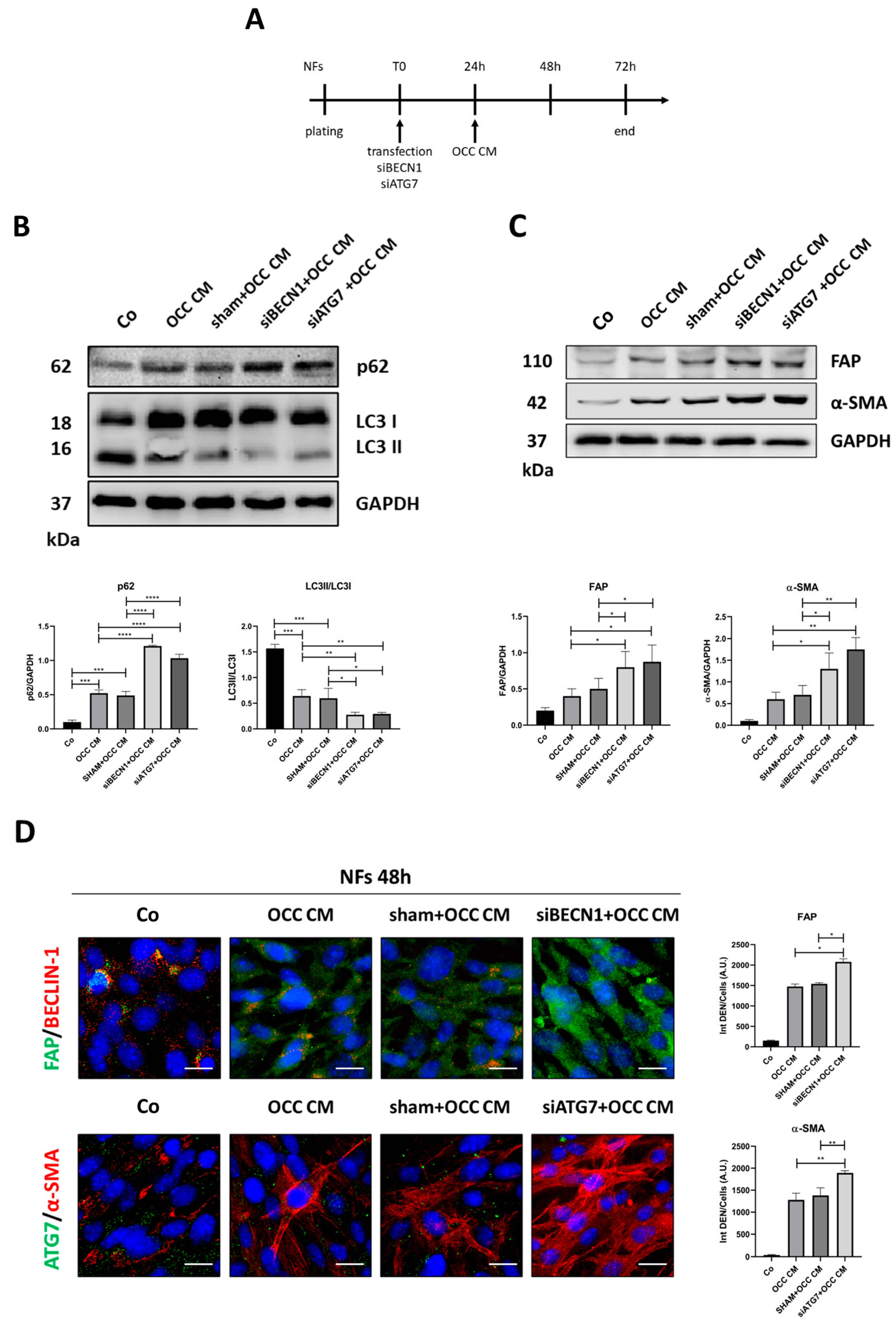
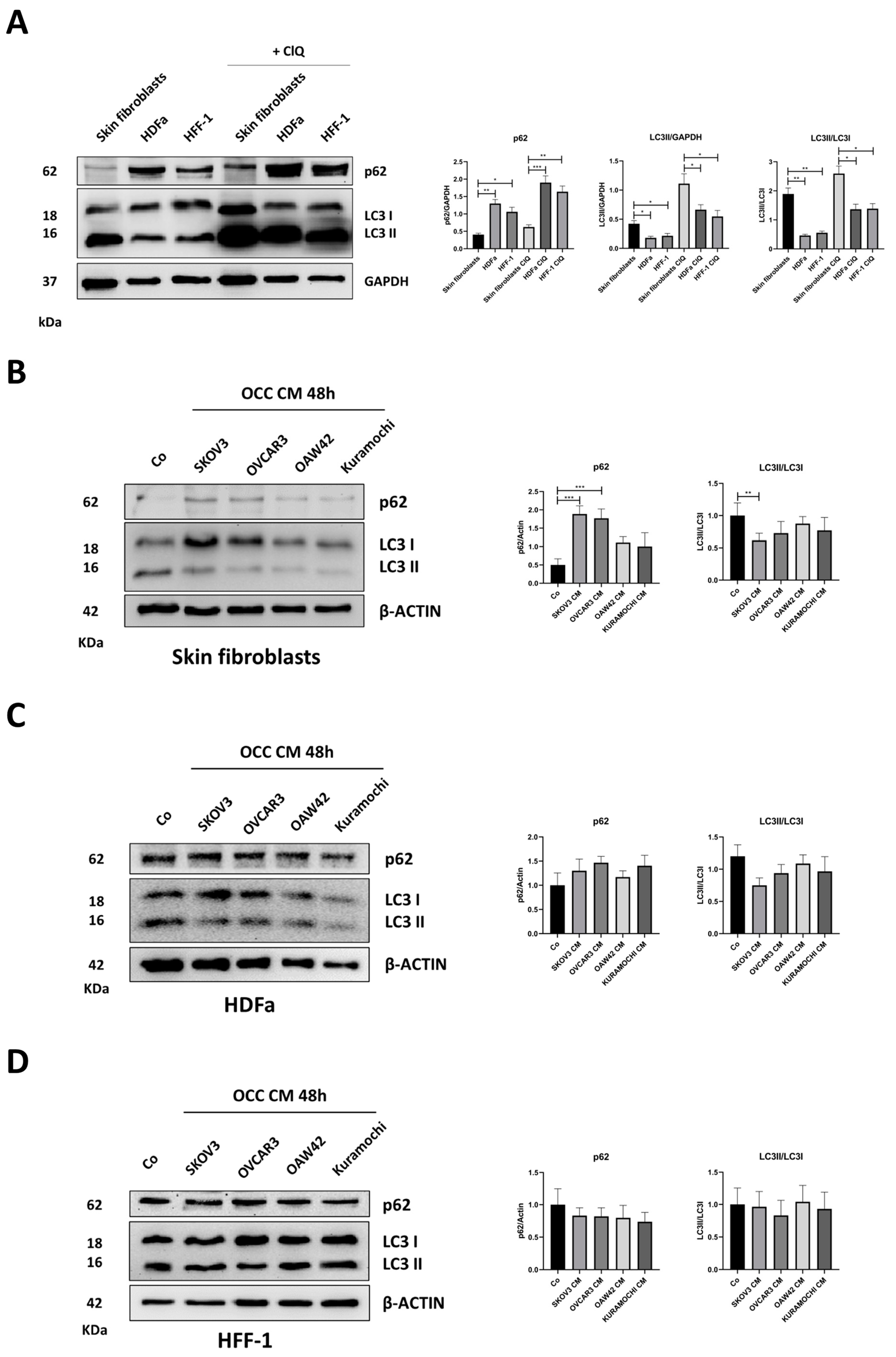
2.3. Autophagy Opposes the Transition towards CAFs
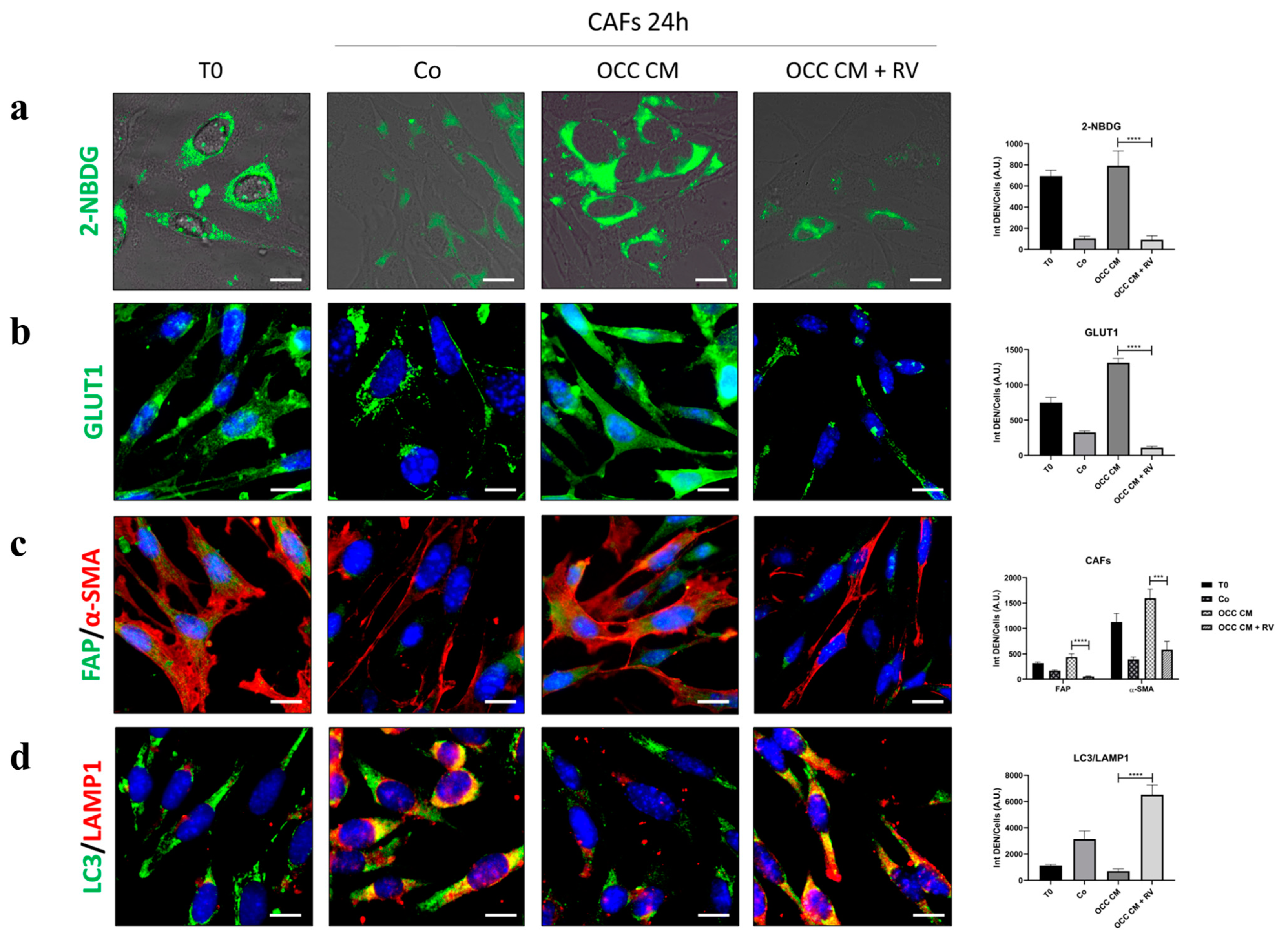
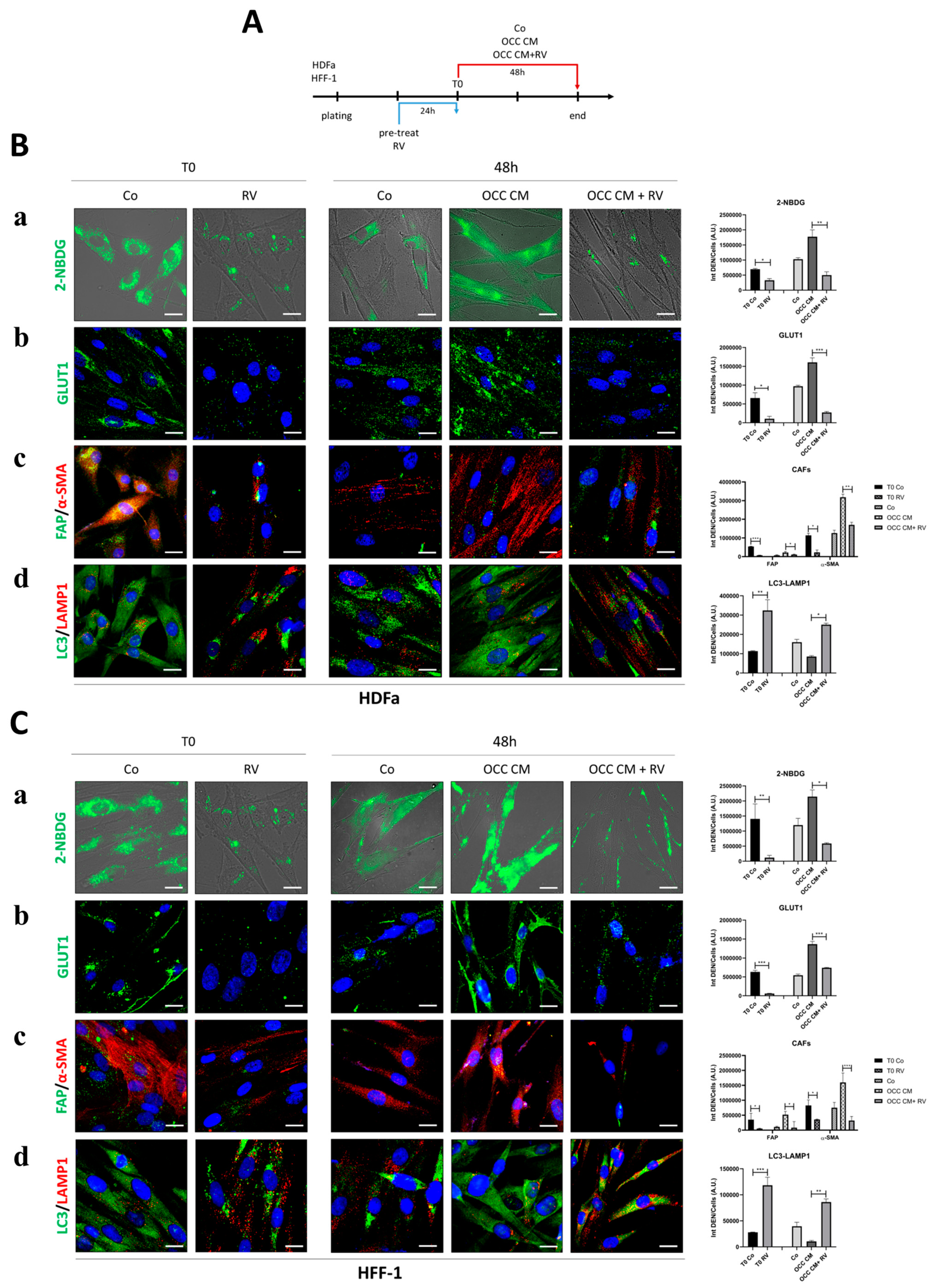
2.4. Resveratrol Reprograms CAFs to NFs
2.5. Autophagy Upregulation Reprograms Myofibroblasts to NFs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Antibodies
4.4. Collection of the Ovarian Cancer Cell-Conditioning Medium (OCC CM)
4.5. Glucose Uptake Assay
4.6. Cell Transfection
4.7. Western Blotting
4.8. Immunofluorescence
4.9. Imaging Acquisition and Analysis
4.10. Co-Immunoprecipitation
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, J.; Chan, W.C.; Ngai, C.H.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E.; Xu, W.; Zheng, Z.-J.; Elcarte, E.; Withers, M.; et al. Worldwide Burden, Risk Factors, and Temporal Trends of Ovarian Cancer: A Global Study. Cancers 2022, 14, 2230. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M. Ovarian Cancer Stroma: Pathophysiology and the Roles in Cancer Development. Cancers 2012, 4, 701–724. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Faget, D.V.; Beachy, P.; Beebe, D.; Bhowmick, N.A.; Cukierman, E.; Deng, W.-M.; Granneman, J.G.; Hildesheim, J.; Kalluri, R.; et al. Deconstructing tumor heterogeneity: The stromal perspective. Oncotarget 2020, 11, 3621–3632. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.A.; Nilsson, E.; Mosher, R.; Magrane, G.; Albertson, D.; Pinkel, D.; Gray, J.W.; Skinner, M.K. Stromal-epithelial interactions in the progression of ovarian cancer: Influence and source of tumor stromal cells. Mol. Cell. Endocrinol. 2001, 175, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2, e92232. [Google Scholar] [CrossRef]
- Quiros, R.M.; Valianou, M.; Kwon, Y.; Brown, K.M.; Godwin, A.K.; Cukierman, E. Ovarian normal and tumor-associated fibroblasts retain in vivo stromal characteristics in a 3-D matrix-dependent manner. Gynecol. Oncol. 2008, 110, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef] [PubMed]
- Louault, K.; Li, R.-R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Haviv, I.; Polyak, K.; Qiu, W.; Hu, M.; Campbell, I. Origin of carcinoma associated fibroblasts. Cell Cycle 2009, 8, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shi, X.; Yang, M.; Luo, J.; Gao, Q.; Wang, X.; Wu, Y.; Tian, Y.; Wu, F.; Zhou, H. Glycolysis reprogramming in cancer-associated fibroblasts promotes the growth of oral cancer through the lncRNA H19/miR-675-5p/PFKFB3 signaling pathway. Int. J. Oral Sci. 2021, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Li, W.; Li, X.; Jin, X.; Liao, Q.; Li, Y.; Zhou, Y. ‘Reverse Warburg effect’ of cancer associated fibroblasts (Review). Int. J. Oncol. 2022, 60, 67. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Girone, C.; Esposito, A.; Vidoni, C.; Vallino, L.; Secomandi, E.; Dhanasekaran, D.N.; Isidoro, C. How Autophagy Shapes the Tumor Microenvironment in Ovarian Cancer. Front. Oncol. 2020, 10, 599915. [Google Scholar] [CrossRef]
- Thuwajit, C.; Ferraresi, A.; Titone, R.; Thuwajit, P.; Isidoro, C. The metabolic cross-talk between epithelial cancer cells and stromal fibroblasts in ovarian cancer progression: Autophagy plays a role. Med. Res. Rev. 2018, 38, 1235–1254. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lan, H.; He, D.; Wang, Z.; Xu, R.; Yuan, J.; Xiao, M.; Zhang, Y.; Gong, L.; Xiao, S.; et al. Analysis of Autophagy-Related Signatures Identified Two Distinct Subtypes for Evaluating the Tumor Immune Microenvironment and Predicting Prognosis in Ovarian Cancer. Front. Oncol. 2021, 11, 616133. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ding, J.; Wang, C.; Sun, Y.; Guo, J.; Liu, S.; Cheng, Z. Identification of an Autophagy-Related Signature for Prognosis and Immunotherapy Response Prediction in Ovarian Cancer. Biomolecules 2023, 13, 339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sari, D.; Gozuacik, D.; Akkoc, Y. Role of autophagy in cancer-associated fibroblast activation, signaling and metabolic reprograming. Front. Cell Dev. Biol. 2024, 11, 1274682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. BioMed Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumor Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vidoni, C.; Ferraresi, A.; Vallino, L.; Salwa, A.; Ha, J.H.; Seca, C.; Garavaglia, B.; Dhanasekaran, D.N.; Isidoro, C. Glycolysis Inhibition of Autophagy Drives Malignancy in Ovarian Cancer: Exacerbation by IL-6 and Attenuation by Resveratrol. IJMS 2023, 24, 1723. [Google Scholar] [CrossRef]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef] [PubMed]
- Follo, C.; Vidoni, C.; Morani, F.; Ferraresi, A.; Seca, C.; Isidoro, C. Amino acid response by Halofuginone in Cancer cells triggers autophagy through proteasome degradation of mTOR. Cell Commun. Signal 2019, 17, 39. [Google Scholar] [CrossRef]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Titone, R.; Follo, C.; Castiglioni, A.; Chiorino, G.; Dhanasekaran, D.N.; Isidoro, C. The protein restriction mimetic Resveratrol is an autophagy inducer stronger than amino acid starvation in ovarian cancer cells. Mol. Carcinog. 2017, 56, 2681–2691. [Google Scholar] [CrossRef]
- Vallino, L.; Ferraresi, A.; Vidoni, C.; Secomandi, E.; Esposito, A.; Dhanasekaran, D.N.; Isidoro, C. Modulation of non-coding RNAs by resveratrol in ovarian cancer cells: In silico analysis and literature review of the anti-cancer pathways involved. J. Tradit. Complement. Med. 2020, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Savio, M.; Ferraresi, A.; Corpina, C.; Vandenberghe, S.; Scarlata, C.; Sottile, V.; Morini, L.; Garavaglia, B.; Isidoro, C.; Stivala, L.A. Resveratrol and Its Analogue 4,4′-Dihydroxy-trans-stilbene Inhibit Lewis Lung Carcinoma Growth In Vivo through Apoptosis, Autophagy and Modulation of the Tumour Microenvironment in a Murine Model. Biomedicines 2022, 10, 1784. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Ferraresi, A.; Vidoni, C.; Loilome, W.; Yongvanit, P.; Namwat, N.; Isidoro, C. Resveratrol interrupts the pro-invasive communication between cancer associated fibroblasts and cholangiocarcinoma cells. Cancer Lett. 2018, 430, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Trope, C.G.; Reich, R. The Role of the Tumor Stroma in Ovarian Cancer. Front. Oncol. 2014, 4, 104. [Google Scholar] [CrossRef] [PubMed]
- Labiche, A.; Heutte, N.; Herlin, P.; Chasle, J.; Gauduchon, P.; Elie, N. Stromal Compartment as a Survival Prognostic Factor in Advanced Ovarian Carcinoma. Int. J. Gynecol. Cancer 2010, 20, 28–33. [Google Scholar] [CrossRef]
- Kochetkova, M.; Samuel, M.S. Differentiation of the tumor microenvironment: Are CAFs the Organizer? Trends Cell Biol. 2022, 32, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Salhia, B. Cancer-Associated Fibroblast Subpopulations with Diverse and Dynamic Roles in the Tumor Microenvironment. Mol. Cancer Res. 2022, 20, 183–192. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Maia, A.; Wiemann, S. Cancer-Associated Fibroblasts: Implications for Cancer Therapy. Cancers 2021, 13, 3526. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Yang, H.; Liang, T.; Bai, X. The “Self-eating” of cancer-associated fibroblast: A potential target for cancer. Biomed. Pharmacother. 2023, 163, 114762. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Gomes, R.N.; Manuel, F.; Nascimento, D.S. The bright side of fibroblasts: Molecular signature and regenerative cues in major organs. Npj Regen. Med. 2021, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Hsia, L.; Ashley, N.; Ouaret, D.; Wang, L.M.; Wilding, J.; Bodmer, W.F. Myofibroblasts are distinguished from activated skin fibroblasts by the expression of AOC3 and other associated markers. Proc. Natl. Acad. Sci. USA 2016, 113, E2162–E2171. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs Reprogram Normal Fibroblasts into Cancer-Associated Fibroblasts in Ovarian Cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Esposito, A.; Ferraresi, A.; Salwa, A.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers 2022, 14, 2142. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Esposito, A.; Girone, C.; Vallino, L.; Salwa, A.; Ghezzi, I.; Thongchot, S.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts LPA-Induced Ovarian Cancer Cell Migration and Platinum Resistance by Rescuing Hedgehog-Mediated Autophagy. Cells 2021, 10, 3213. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy: Resveratrol Inhibits Il-6-Induced Ovarian Cancer Cell Migration. Mol. Carcinog. 2017, 56, 1164–1181. [Google Scholar] [CrossRef]
- Akkoc, Y.; Dalci, K.; Karakas, H.E.; Erbil-Bilir, S.; Yalav, O.; Sakman, G.; Celik, F.; Arikan, S.; Zeybek, U.; Ergin, M.; et al. Tumor-derived CTF1 (cardiotrophin 1) is a critical mediator of stroma-assisted and autophagy-dependent breast cancer cell migration, invasion and metastasis. Autophagy 2023, 19, 306–323. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Clocchiatti, A.; Dotto, G.P. A role for stromal autophagy in cancer-associated fibroblast activation. Autophagy 2019, 15, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.L.; Parkinson, E.K.; Yap, L.F.; Paterson, I.C. Autophagy is deregulated in cancer-associated fibroblasts from oral cancer and is stimulated during the induction of fibroblast senescence by TGF-β1. Sci. Rep. 2021, 11, 584. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II Positively Regulates Glucose Starvation-Induced Autophagy through TORC1 Inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Savoia, P.; Raina, G.; Camillo, L.; Farruggio, S.; Mary, D.; Veronese, F.; Graziola, F.; Zavattaro, E.; Tiberio, R.; Grossini, E. Anti-oxidative effects of 17 β-estradiol and genistein in human skin fibroblasts and keratinocytes. J. Dermatol. Sci. 2018, 92, 62–77. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | BECN1 | TP53 | PTEN | AKT | PI3KCA |
|---|---|---|---|---|---|
| SKOV3 | Wild-type | Null | Wild-type | Wild-type | Mutated |
| OVCAR3 | Wild-type | Mutated | Null | Wild-type | Wild-type |
| OAW42 | Wild-type | Wild-type | Wild-type | Wild-type | Mutated |
| Kuramochi | Wild-type | Mutated | Wild-type | Mutated | Wild-type |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraresi, A.; Girone, C.; Maheshwari, C.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy. Int. J. Mol. Sci. 2024, 25, 5691. https://doi.org/10.3390/ijms25115691
Ferraresi A, Girone C, Maheshwari C, Vallino L, Dhanasekaran DN, Isidoro C. Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy. International Journal of Molecular Sciences. 2024; 25(11):5691. https://doi.org/10.3390/ijms25115691
Chicago/Turabian StyleFerraresi, Alessandra, Carlo Girone, Chinmay Maheshwari, Letizia Vallino, Danny N. Dhanasekaran, and Ciro Isidoro. 2024. "Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy" International Journal of Molecular Sciences 25, no. 11: 5691. https://doi.org/10.3390/ijms25115691
APA StyleFerraresi, A., Girone, C., Maheshwari, C., Vallino, L., Dhanasekaran, D. N., & Isidoro, C. (2024). Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy. International Journal of Molecular Sciences, 25(11), 5691. https://doi.org/10.3390/ijms25115691