
A Causal Regulation Modeling Algorithm for Temporal Events with Application to Escherichia coli’s Aerobic to Anaerobic Transition

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
2.1. Overview of This Study
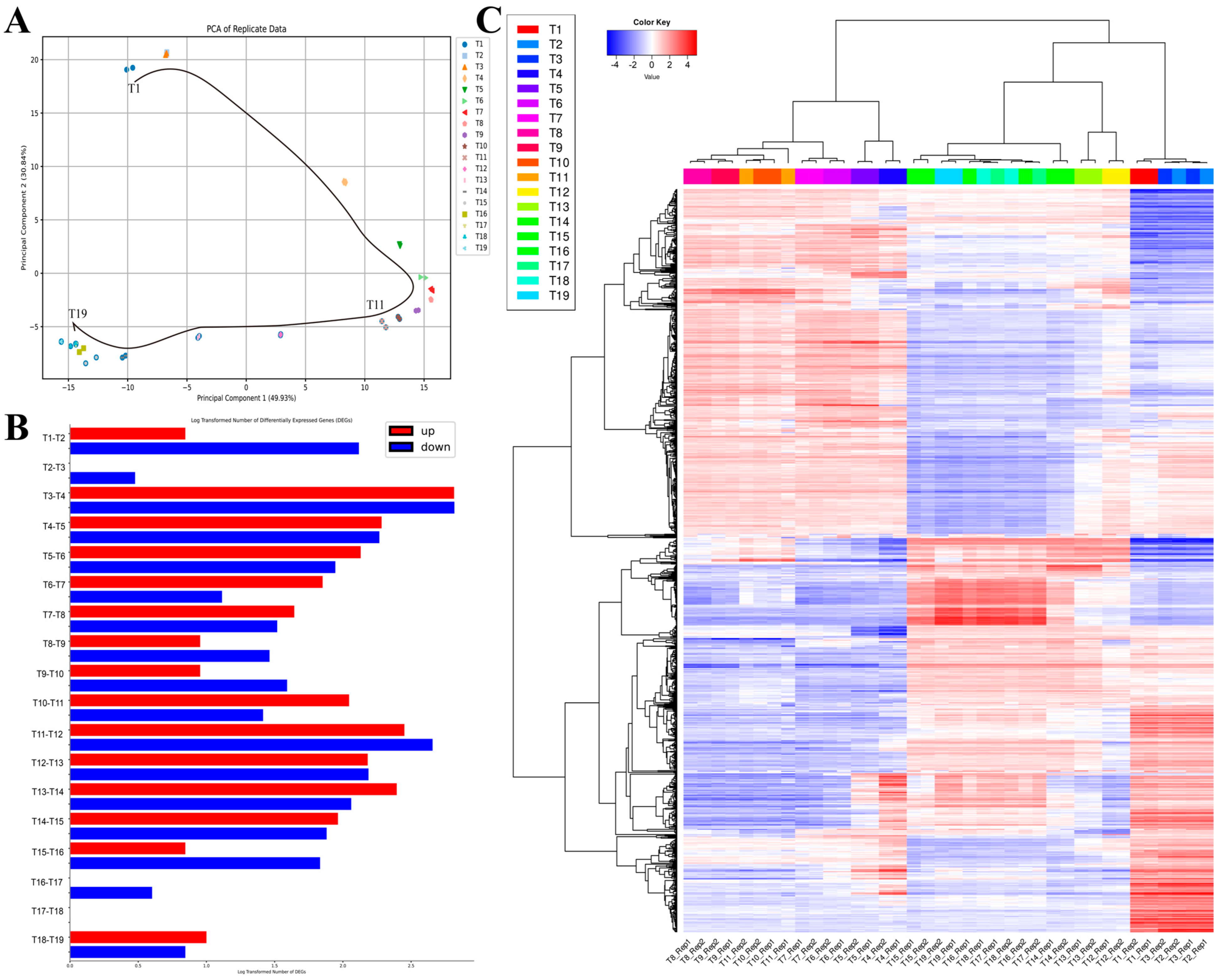
2.2. The Basic Temporal DEG- and Clustering-Based Functional Analysis of E. coli’s AAT Process
2.3. The Regulatory Network Model of E. coli
2.4. Elucidating the Dynamic Regulatory Network in AAT Process through Computational Modeling
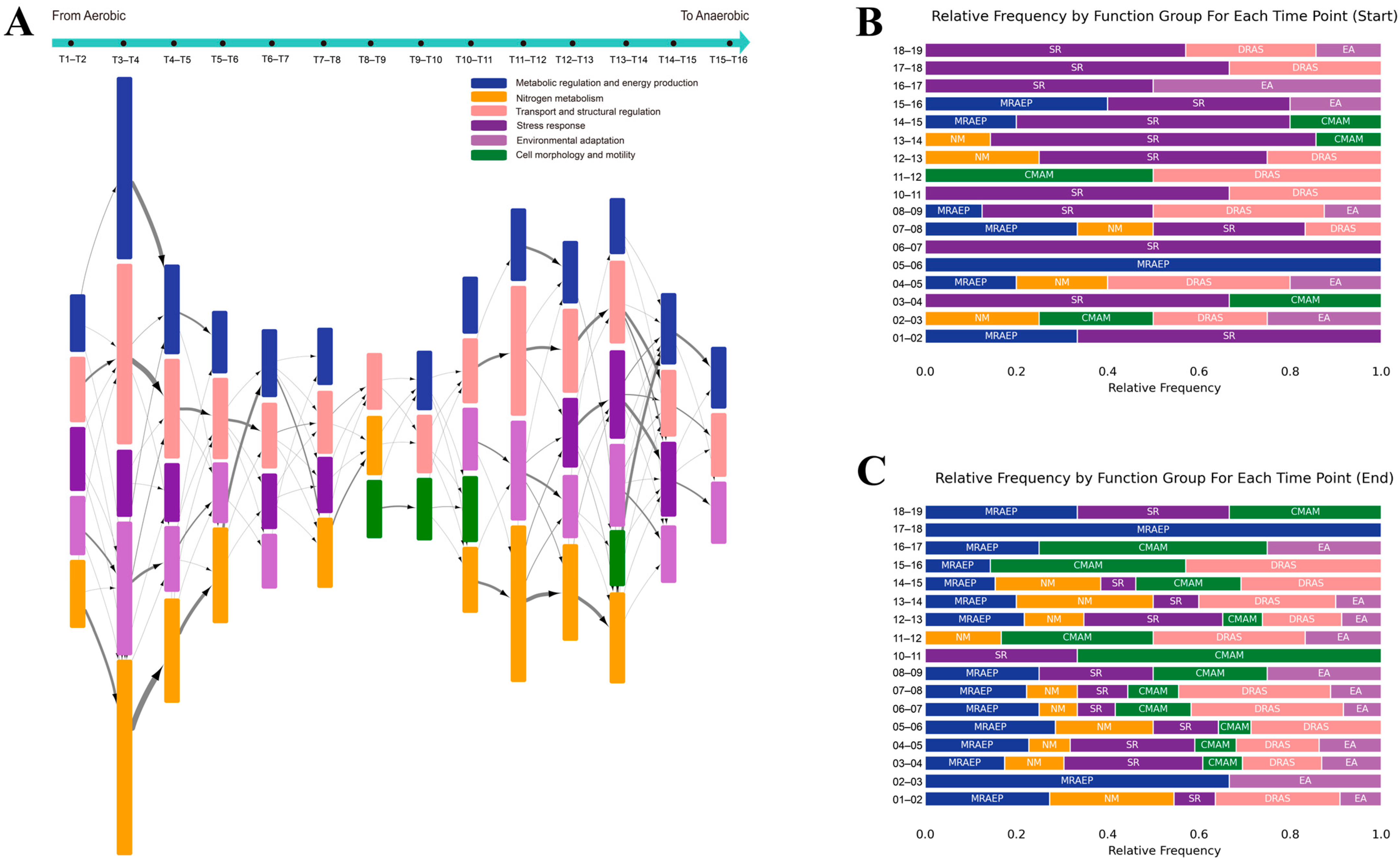
2.5. Visualization of the Signal Transduction in AAT Process by Hypernode Enrichment
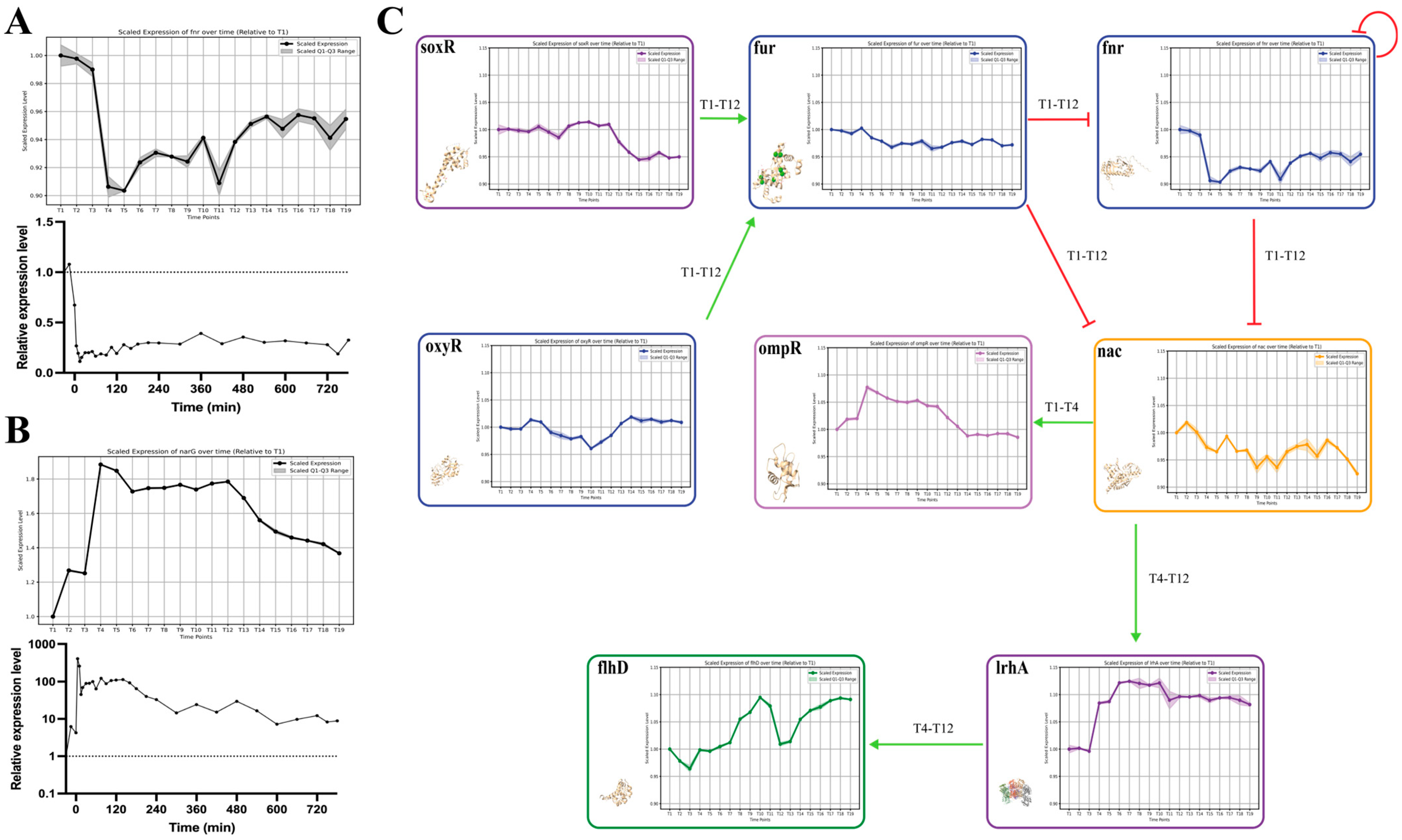
2.6. Validation and Dynamic Analysis of the AAT Regulatory Network through Gene Expression Profiling
3. Discussion
4. Methods and Materials
4.1. Microarray and qRT-PCR Gene Expression Experiments
4.2. E. coli Regulatory Network Model Construction
4.3. Gene Expression Analysis Workflow
4.4. Gene Ontology Information Flow Network
4.5. Causal Signaling Model Algorithm
| Algorithm 1: Primary regulatory drivers dysregulation analysis. |
Inputs:
Timepoint-dependent dysregulating gene lists, along with their associated p-values, revealing insights into the temporal gene regulatory patterns and overarching regulators. |
| Algorithm 2: Enhanced Fisher’s exact test (FET)-based regulatory impact analysis. |
Inputs:
A list of regulators, each associated with its pseudo activation state and corresponding p-value, providing insights into their potential regulatory impact based on the DEG patterns observed within the PKN. |
4.6. Temporal Path Searching
| Algorithm 3: Temporal regulation analysis for identifying key regulatory nodes. |
Procedure:
|
4.7. Hypergraph Generation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arbeitman, M.N.; Furlong, E.E.; Imam, F.; Johnson, E.; Null, B.H.; Baker, B.S.; Krasnow, M.A.; Scott, M.P.; Davis, R.W.; White, K.P. Gene expression during the life cycle of Drosophila melanogaster. Science 2002, 297, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Spellman, P.T.; Sherlock, G.; Zhang, M.Q.; Iyer, V.R.; Anders, K.; Eisen, M.B.; Brown, P.O.; Botstein, D.; Futcher, B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 1998, 9, 3273–3297. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, K.; Salama, N.R.; Tompkins, L.S.; Falkow, S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15136–15141. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.; Hughes, J.D.; Campbell, M.J.; Cho, R.J.; Church, G.M. Systematic determination of genetic network architecture. Nat. Genet. 1999, 22, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, P.; Slonim, D.; Mesirov, J.; Zhu, Q.; Kitareewan, S.; Dmitrovsky, E.; Lander, E.S.; Golub, T.R. Interpreting patterns of gene expression with self-organizing maps. methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Dolled-Filhart, M.; Lin, J.; Yu, H.; Gerstein, M. Beyond synexpression relationships: Local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions. J. Mol. Biol. 2001, 314, 1053–1066. [Google Scholar] [CrossRef]
- Mukhopadhyay, N.D.; Chatterjee, S. Causality and pathway search in microarray time series experiment. Bioinformatics 2007, 23, 442–449. [Google Scholar] [CrossRef]
- Mathavan, S.; Lee, S.G.; Mak, A.; Miller, L.D.; Murthy, K.R.; Govindarajan, K.R.; Tong, Y.; Wu, Y.L.; Lam, S.H.; Yang, H.; et al. Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet. 2005, 1, 260–276. [Google Scholar] [CrossRef]
- Marku, M.; Pancaldi, V. From time-series transcriptomics to gene regulatory networks: A review on inference methods. PLOS Comput. Biol. 2023, 19, e1011254. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Rodriguez, M.; Zirngibl, K.; Ivanova, O.; Lobentanzer, S.; Saez-Rodriguez, J. Integrating knowledge and omics to decipher mechanisms via large-scale models of signaling networks. Mol. Syst. Biol. 2022, 18, e11036. [Google Scholar] [CrossRef] [PubMed]
- Bradley, G.; Barrett, S.J. CausalR: Extracting mechanistic sense from genome scale data. Bioinformatics 2017, 33, 3670–3672. [Google Scholar] [CrossRef] [PubMed]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: An empirical gene set approach exploiting pathway topology. Nucleic Acids Res. 2013, 41, e19. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shen, L.; Shang, X.; Liu, W. Subpathway Analysis based on Signaling-Pathway Impact Analysis of Signaling Pathway. PLoS ONE 2015, 10, e0132813. [Google Scholar] [CrossRef] [PubMed]
- Köksal, A.S.; Beck, K.; Cronin, D.R.; McKenna, A.; Camp, N.D.; Srivastava, S.; MacGilvray, M.E.; Bodík, R.; Wolf-Yadlin, A.; Fraenkel, E.; et al. Synthesizing Signaling Pathways from Temporal Phosphoproteomic Data. Cell Rep. 2018, 24, 3607–3618. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Trairatphisan, P.; Gjerga, E.; Didangelos, A.; Barratt, J.; Saez-Rodriguez, J. From expression footprints to causal pathways: Contextualizing large signaling networks with CARNIVAL. NPJ Syst. Biol. Appl. 2019, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 2010, 20, 5–14. [Google Scholar] [CrossRef]
- Partridge, J.D.; Sanguinetti, G.; Dibden, D.P.; Roberts, R.E.; Poole, R.K.; Green, J. Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J. Biol. Chem. 2007, 282, 11230–11237. [Google Scholar] [CrossRef]
- Sousa, P.M.F.; Videira, M.A.M.; Bohn, A.; Hood, B.L.; Conrads, T.P.; Goulao, L.F.; Melo, A.M.P. The aerobic respiratory chain of Escherichia coli: From genes to supercomplexes. Microbiology 2012, 158 Pt 9, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- von Wulffen, J.; RecogNice, T.; Sawodny, O.; Feuer, R. Transition of an Anaerobic Escherichia coli Culture to Aerobiosis: Balancing mRNA and Protein Levels in a Demand-Directed Dynamic Flux Balance Analysis. PLoS ONE 2016, 11, e0158711. [Google Scholar] [CrossRef] [PubMed]
- Partridge, J.D.; Scott, C.; Tang, Y.; Poole, R.K.; Green, J. Escherichia coli transcriptome dynamics during the transition from anaerobic to aerobic conditions. J. Biol. Chem. 2006, 281, 27806–27815. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, C.R.; Decker, K.T.; Sastry, A.V.; Rychel, K.; Gao, Y.; McConn, J.L.; Zielinski, D.C.; Palsson, B.O. A multi-scale expression and regulation knowledge base for Escherichia coli. Nucleic Acids Res. 2023, 51, 10176–10193. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, R.P.; Park, S.J. Aerobic-anaerobic gene regulation in Escherichia coli: Control by the ArcAB and Fnr regulons. Res. Microbiol. 1994, 145, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.T.; Selvarajoo, K. Attractor Concepts to Evaluate the Transcriptome-wide Dynamics Guiding Anaerobic to Aerobic State Transition in Escherichia coli. Sci. Rep. 2020, 10, 5878. [Google Scholar] [CrossRef] [PubMed]
- Sima, C.; Hua, J.; Jung, S. Inference of gene regulatory networks using time-series data: A survey. Curr. Genom. 2009, 10, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Singh, P.; Seshasayee, A.S.N. Understanding the Genome-Wide Transcription Response to Various cAMP Levels in Bacteria Using Phenomenological Models. mSystems 2022, 7, e0090022. [Google Scholar] [CrossRef]
- Benedetti, E.; Pucic-Bakovic, M.; Keser, T.; Gerstner, N.; Buyukozkan, M.; Stambuk, T.; Selman, M.H.J.; Rudan, I.; Polasek, O.; Hayward, C.; et al. A strategy to incorporate prior knowledge into correlation network cutoff selection. Nat. Commun. 2020, 11, 5153. [Google Scholar] [CrossRef]
- Erbe, R.; Gore, J.; Gemmill, K.; Gaykalova, D.A.; Fertig, E.J. The use of machine learning to discover regulatory networks controlling biological systems. Mol. Cell 2022, 82, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Dorier, J.; Crespo, I.; Niknejad, A.; Liechti, R.; Ebeling, M.; Xenarios, I. Boolean regulatory network reconstruction using literature based knowledge with a genetic algorithm optimization method. BMC Bioinform. 2016, 17, 410. [Google Scholar] [CrossRef] [PubMed]
- Silvela, J.; Portillo, J. Breadth-first search and its application to image processing problems. IEEE Trans. Image Process 2001, 10, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Weber, K.D.; Qiu, Y.; Kiley, P.J.; Blattner, F.R. Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 2005, 187, 1135–1160. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martinez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muniz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Paley, S.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Moore, L.R.; Subhraveti, P.; Gama-Castro, S.; Tierrafria, V.H.; et al. The EcoCyc Database (2023). EcoSal Plus 2023, 11, eesp00022023. [Google Scholar] [CrossRef] [PubMed]
- Salgado, H.; Martinez-Flores, I.; Bustamante, V.H.; Alquicira-Hernandez, K.; Garcia-Sotelo, J.S.; Garcia-Alonso, D.; Collado-Vides, J. Using RegulonDB, the Escherichia coli K-12 Gene Regulatory Transcriptional Network Database. Curr. Protoc. Bioinform. 2018, 61, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, Y.C.; Luo, Y.; Cai, X.; Qiu, P.; Cui, S.; Wang, Z.; Huang, H.Y.; Huang, H.D. Quantitative model for genome-wide cyclic AMP receptor protein binding site identification and characteristic analysis. Brief. Bioinform. 2023, 24, bbad138. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodriguez Martinez, M.; Lopez, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J.; Tugendreich, S., Jr. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Ritz, A.; Tegge, A.N.; Kim, H.; Poirel, C.L.; Murali, T.M. Signaling hypergraphs. Trends Biotechnol. 2014, 32, 356–362. [Google Scholar] [CrossRef]
- Betzel, R.F. Community detection in network neuroscience. In Connectome Analysis; Elsevier: Amsterdam, The Netherlands, 2023; pp. 149–171. [Google Scholar]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An extendable pathway analysis toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism Type | Number of Edges from EcoCyc | Number of Edges from RegulonDB | Number of Edges from STRING |
|---|---|---|---|
| Gene–Gene Correlation | 0 | 0 | 1,089,246 |
| Transcriptional Regulation | 8199 | 4814 | 0 |
| Protein–Protein Interaction | 569 | 428 | 582 |
| Metabolic Reactions | 5630 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Mao, R.; Xu, J.; Huang, Y.; Xu, J.; Cui, S.; Zhu, Z.; Ji, X.; Huang, S.; Huang, Y.; et al. A Causal Regulation Modeling Algorithm for Temporal Events with Application to Escherichia coli’s Aerobic to Anaerobic Transition. Int. J. Mol. Sci. 2024, 25, 5654. https://doi.org/10.3390/ijms25115654
Chen Y, Mao R, Xu J, Huang Y, Xu J, Cui S, Zhu Z, Ji X, Huang S, Huang Y, et al. A Causal Regulation Modeling Algorithm for Temporal Events with Application to Escherichia coli’s Aerobic to Anaerobic Transition. International Journal of Molecular Sciences. 2024; 25(11):5654. https://doi.org/10.3390/ijms25115654
Chicago/Turabian StyleChen, Yigang, Runbo Mao, Jiatong Xu, Yixian Huang, Jingyi Xu, Shidong Cui, Zihao Zhu, Xiang Ji, Shenghan Huang, Yanzhe Huang, and et al. 2024. "A Causal Regulation Modeling Algorithm for Temporal Events with Application to Escherichia coli’s Aerobic to Anaerobic Transition" International Journal of Molecular Sciences 25, no. 11: 5654. https://doi.org/10.3390/ijms25115654
APA StyleChen, Y., Mao, R., Xu, J., Huang, Y., Xu, J., Cui, S., Zhu, Z., Ji, X., Huang, S., Huang, Y., Huang, H.-Y., Yen, S.-C., Lin, Y.-C.-D., & Huang, H.-D. (2024). A Causal Regulation Modeling Algorithm for Temporal Events with Application to Escherichia coli’s Aerobic to Anaerobic Transition. International Journal of Molecular Sciences, 25(11), 5654. https://doi.org/10.3390/ijms25115654