
Cross-Species Insights into Autosomal Dominant Polycystic Kidney Disease: Provide an Alternative View on Research Advancement




Abstract
1. Introduction
2. Comprehensive Investigation of Multi-Species Epidemiology
3. Pathological Features and Cross-Species Manifestations of ADPKD
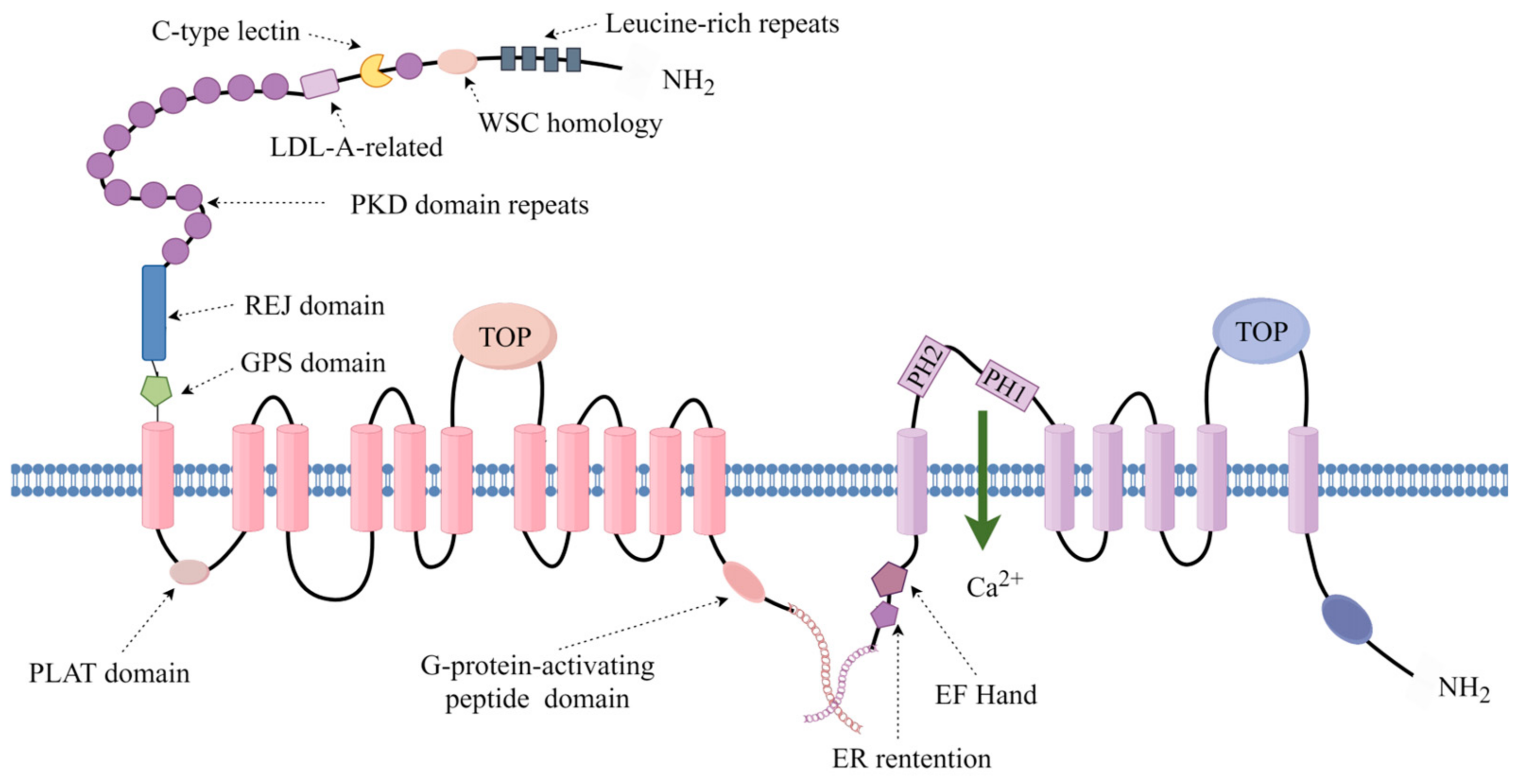
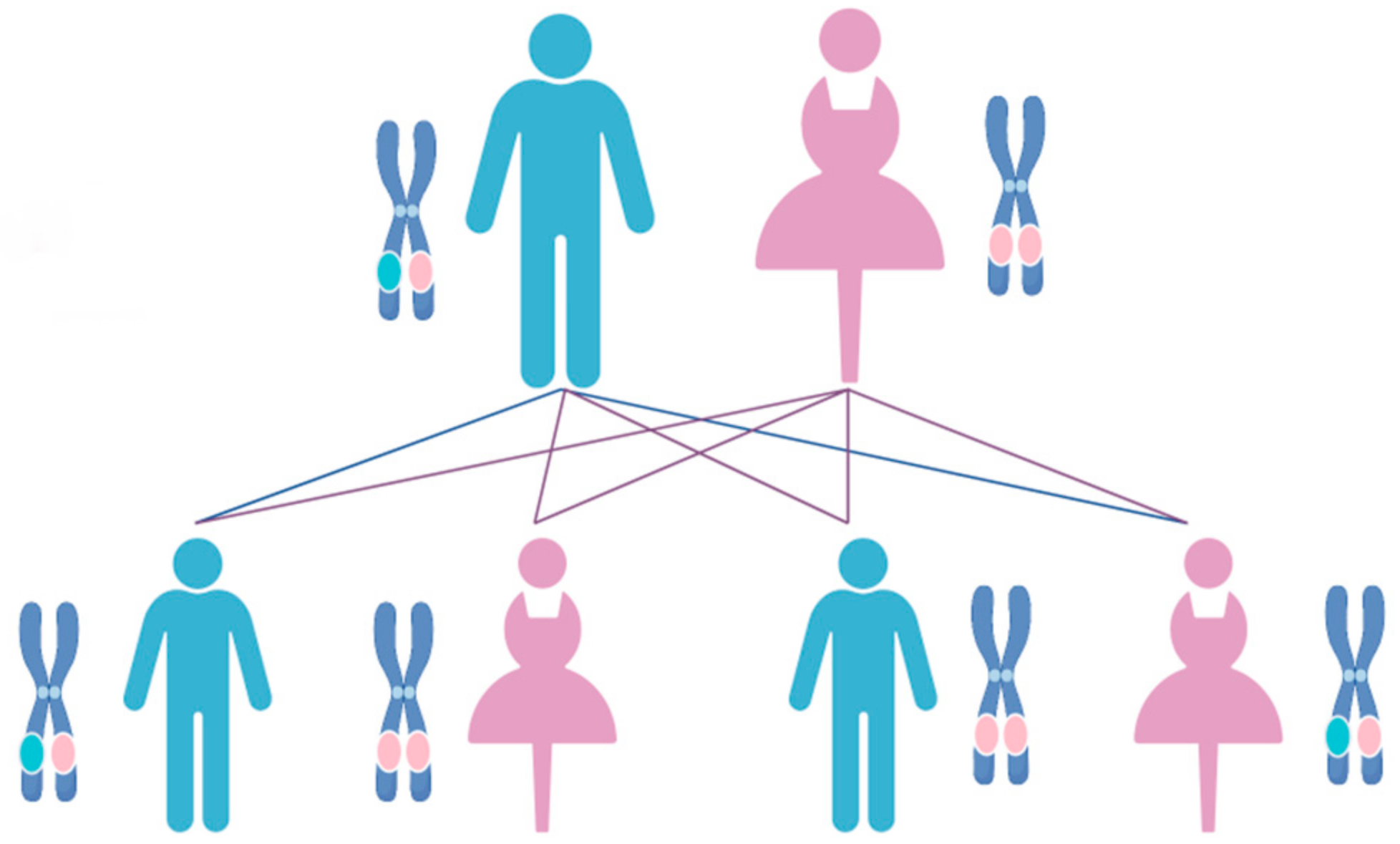
4. Molecular and Genetic Mechanisms of ADPKD across Species
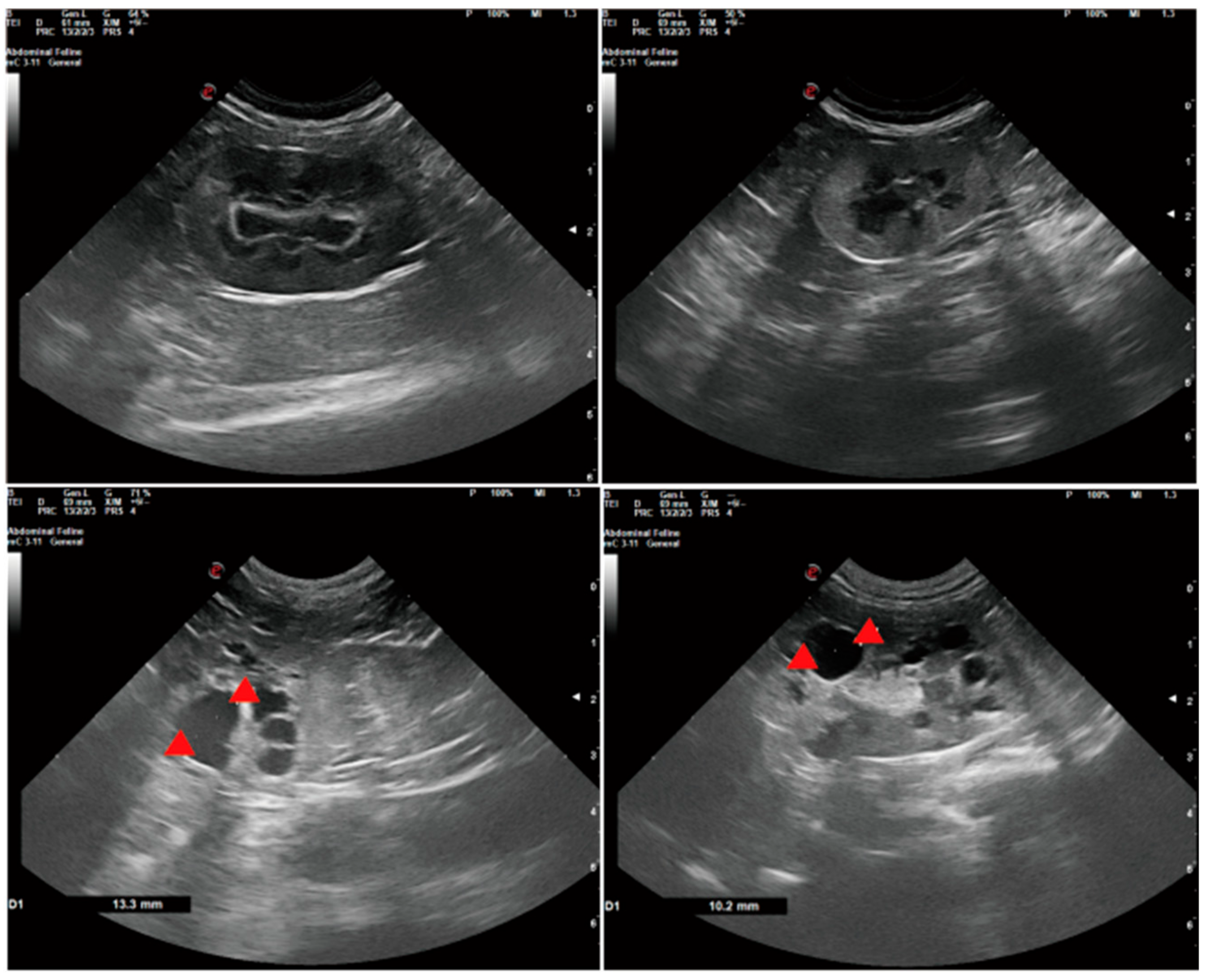
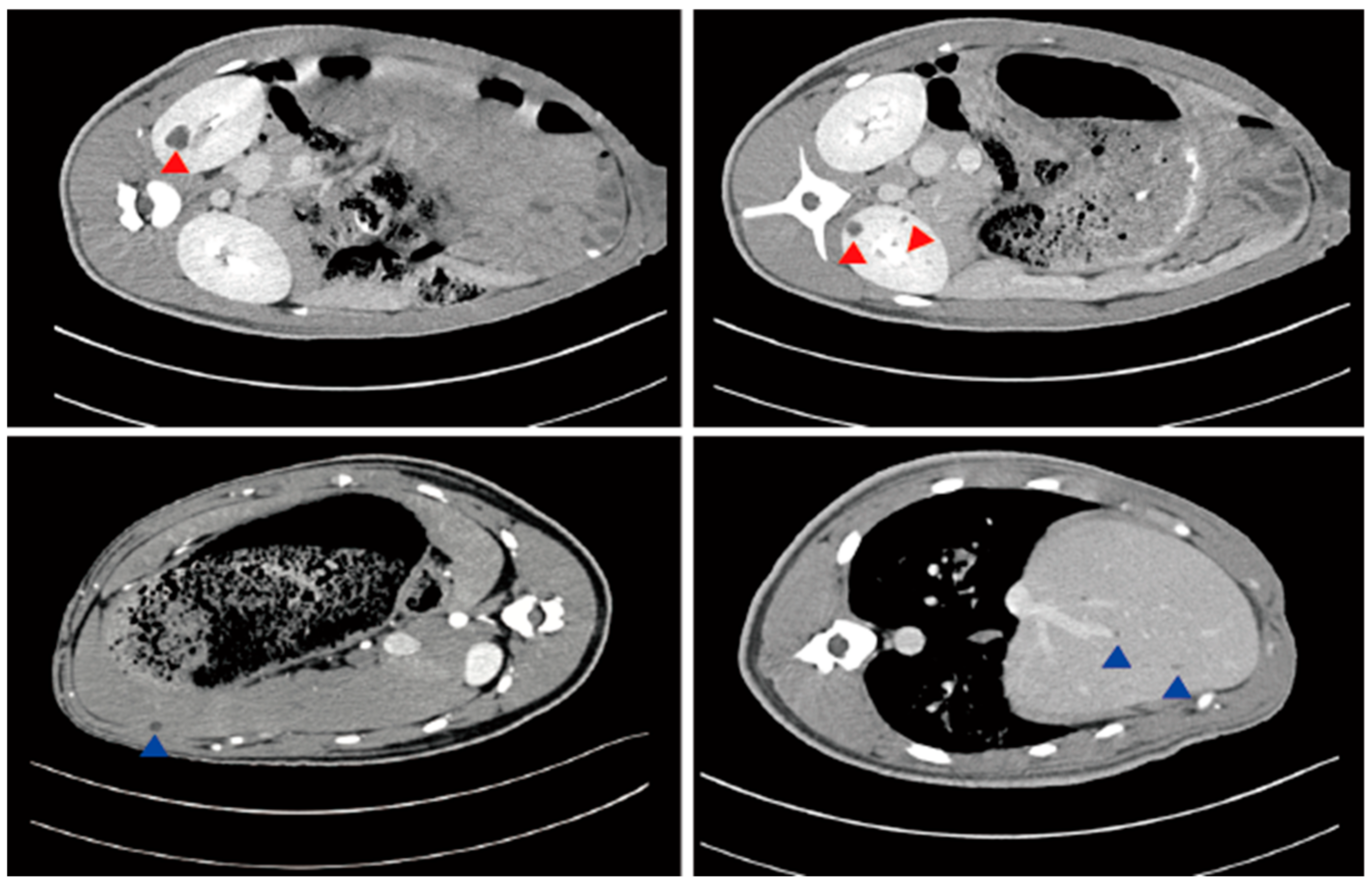
5. Diagnostic Approaches and Clinical Manifestations
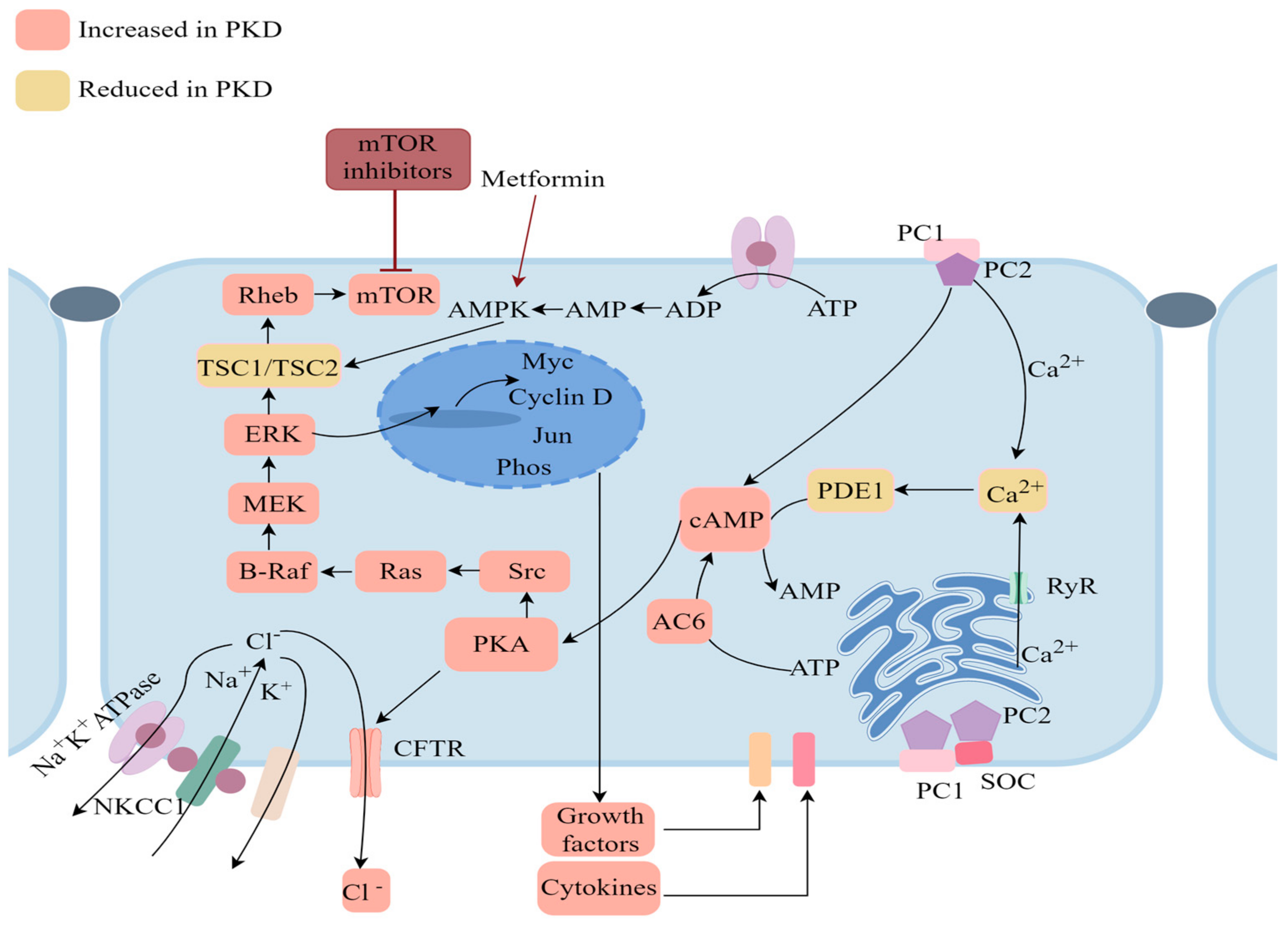
6. Advancements and Challenges in ADPKD Treatment Strategies
7. Advancements in Animal Models for ADPKD Research
8. Conclusions
Funding
Conflicts of Interest
References
- Sutters, M. The pathogenesis of autosomal dominant polycystic kidney disease. Nephron Exp. Nephrol. 2006, 103, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Mochizuki, T.; Muto, S.; Hanaoka, K.; Fukushima, Y.; Narita, I.; Nutahara, K.; Tsuchiya, K.; Tsuruya, K.; Kamura, K.; et al. Evidence-based clinical practice guidelines for polycystic kidney disease. Clin. Exp. Nephrol. 2016, 20, 510. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.U.; Haas, C.S.; Sayer, J.A. Practical approaches to the management of autosomal dominant polycystic kidney disease patients in the era of tolvaptan. Clin. Kidney J. 2018, 11, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2016, 67, 792–810. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Leonhard, W.N.; Zandbergen, M.; Veraar, K.; van den Berg, S.; van der Weerd, L.; Breuning, M.; de Heer, E.; Peters, D.J. Scattered Deletion of PKD1 in Kidneys Causes a Cystic Snowball Effect and Recapitulates Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2015, 26, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, E.J.; Prins, P. Polycystic Kidney Disease (PKD). Tijdschr. Diergeneeskd. 2005, 130, 184–185. [Google Scholar] [PubMed]
- Lowrie, M.; Garosi, L. Classification of involuntary movements in dogs: Paroxysmal dyskinesias. Vet. J. 2017, 220, 65–71. [Google Scholar] [CrossRef]
- O’Leary, C.A.; Duffy, D.; Biros, I.; Corley, S. Linkage confirms canine pkd1 orthologue as a candidate for bull terrier polycystic kidney disease. Anim. Genet. 2009, 40, 543–546. [Google Scholar] [CrossRef]
- Schmidt, U. Angeborene zystische Veränderungen in Leber und Nieren bei einem Kalb [Congenital cystic changes of the liver and kidneys in a calf]. Dtsch. Tierarztl. Wochenschr. 1973, 80, 329. [Google Scholar]
- Patterson, D.S.; Done, J.T. Neurochemistry as a diagnostic aid in the congenital tremor syndrome of piglets. Br. Vet. J. 1977, 133, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Torres, V.E. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Messchendorp, A.L.; Spithoven, E.M.; Casteleijn, N.F.; Dam, W.A.; van den Born, J.; Tonnis, W.F.; Gaillard, C.A.J.M.; Meijer, E. Association of plasma somatostatin with disease severity and progression in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2018, 19, 368. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamed, M.H.; Alsahan, N.; Rice, S.J.; Edwards, N.; Nooreddeen, E.; Alotaibi, M.; Kurdi, W.; Alnemer, M.; Altaleb, N.; Ali, W.; et al. Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr. Nephrol. 2019, 34, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Bai, B.; Li, F.; Bai, R.; Zhuo, Y.; Zhu, Z.; Jia, R.; Li, S.; Chen, Y.; Lan, X. Phenotypes and genetic etiology of spontaneous polycystic kidney and liver disease in cynomolgus monkey. Front. Vet. Sci. 2023, 10, 1106016. [Google Scholar] [CrossRef]
- Mellersh, C.S.; Hitte, C.; Richman, M.; Vignaux, F.; Priat, C.; Jouquand, S.; Werner, P.; André, C.; DeRose, S.; Patterson, D.F.; et al. An integrated linkage-radiation hybrid map of the canine genome. Mamm. Genome 2000, 11, 120–130. [Google Scholar] [CrossRef]
- Sinha, D.; Ivan, D.; Gibbs, E.; Chetluru, M.; Goss, J.; Chen, Q. Fission yeast polycystin Pkd2p promotes cell size expansion and antagonizes the Hippo-related SIN pathway. J. Cell Sci. 2022, 135, 259046. [Google Scholar] [CrossRef]
- Weimbs, T. Polycystic kidney disease and renal injury repair: Common pathways, fluid flow, and the function of polycystin-1. Am. J. Physiol. Ren. Physiol. 2007, 293, F1423–F1432. [Google Scholar] [CrossRef]
- Weimbs, T. Third-hit signaling in renal cyst formation. J. Am. Soc. Nephrol. 2011, 22, 793–795. [Google Scholar] [CrossRef]
- Raman, A.; Parnell, S.C.; Zhang, Y.; Reif, G.A.; Dai, Y.; Khanna, A.; Daniel, E.; White, C.; Vivian, J.L.; Wallace, D.P. Periostin overexpression in collecting ducts accelerates renal cyst growth and fibrosis in polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2018, 315, 1695–1707. [Google Scholar] [CrossRef]
- Raman, A.; Reif, G.A.; Dai, Y.; Khanna, A.; Li, X.; Astleford, L.; Parnell, S.C.; Calvet, J.P.; Wallace, D.P. Integrin-Linked Kinase Signaling Promotes Cyst Growth and Fibrosis in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2708–2719. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Hunter, L.W.; Li, M.; Marin-Padilla, M.; Prakash, Y.S.; Somlo, S.; Harris, P.C.; Torres, V.E.; Sieck, G.C. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum. Mol. Genet. 2003, 12, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Peters, D.J.M.; Klanke, B.; Weidemann, A.; Willam, C.; Schley, G.; Kunzelmann, K.; Eckardt, K.U.; Buchholz, B. HIF-1α promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 2018, 94, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Edelstein, C.L. Apoptosis and autophagy in polycystic kidney disease (PKD). Cell. Signal. 2020, 68, 109518. [Google Scholar] [CrossRef]
- Sekine, A.; Hidaka, S.; Moriyama, T.; Shikida, Y.; Shimazu, K.; Ishikawa, E.; Uchiyama, K.; Kataoka, H.; Kawano, H.; Kurashige, M.; et al. Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). J. Clin. Med. 2022, 11, 6528. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Song, C.J.; Zimmerman, K.A.; Henke, S.J.; Yoder, B.K. Inflammation and Fibrosis in Polycystic Kidney Disease. Results Probl. Cell Differ. 2017, 60, 323–344. [Google Scholar] [PubMed]
- Yuan, J.; Rozengurt, E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J. Cell Biochem. 2008, 103, 648–662. [Google Scholar] [CrossRef]
- Antignac, C.; Calvet, J.P.; Germino, G.G.; Grantham, J.J.; Guay-Woodford, L.M.; Harris, P.C.; Hildebrandt, F.; Peters, D.J.; Somlo, S.; Torres, V.E.; et al. The Future of Polycystic Kidney Disease Research—As Seen By the 12 Kaplan Awardees. J. Am. Soc. Nephrol. 2015, 26, 2081–2095. [Google Scholar] [CrossRef]
- Lakhia, R.; Mishra, A.; Biggers, L.; Malladi, V.; Cobo-Stark, P.; Hajarnis, S.; Patel, V. Enhancer and super-enhancer landscape in polycystic kidney disease. Kidney Int. 2023, 103, 87–99. [Google Scholar] [CrossRef]
- Magistroni, R.; Boletta, A. Defective glycolysis and the use of 2-deoxy-D-glucose in polycystic kidney disease: From animal models to humans. J. Nephrol. 2017, 30, 511–519. [Google Scholar] [CrossRef]
- Nowak, K.L.; Hopp, K. Metabolic Reprogramming in Autosomal Dominant Polycystic Kidney Disease: Evidence and Therapeutic Potential. Clin. J. Am. Soc. Nephrol. 2020, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.; Gedroyc, W.; Hill, P. ADPKD-what the radiologist should know. Br. J. Radiol. 2019, 92, 20190078. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Zhou, C.C.; Wu, M.; Mei, C.L. The Clinical Manifestation and Management of Autosomal Dominant Polycystic Kidney Disease in China. Kidney Dis. 2016, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Pippias, M.; Kramer, A.; Noordzij, M.; Afentakis, N.; Alonso de la Torre, R.; Ambühl, P.M.; Aparicio Madre, M.I.; Arribas Monzón, F.; Åsberg, A.; Bonthuis, M.; et al. The European Renal Association—European Dialysis and Transplant Association Registry Annual Report 2014: A summary. Clin. Kidney J. 2017, 10, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Laruelle, E.; Couchoud, C.; Collart, F.; Cases, A.; Arici, M.; Helve, J.; Waldum-Grevbo, B.; et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014, 86, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Lulich, J.P.; Osborne, C.A.; Walter, P.A. O’Brien TD:Feline idiopathic polycystic kidney disease. Compend. Cont. Ed. Pract. Vet. 1988, 10, 1029–1041. [Google Scholar]
- Podell, M.; DiBartola, S.P.; Rosol, T.J. Polycystic kidney disease and renal lymphoma in a cat. J. Am. Vet. Med. Assoc. 1992, 201, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, K.E. Polycystic disease of the kidney and liver in an adult Persian cat. J. Comp. Pathol. 1989, 100, 327–330. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Kumar, S.; Gabow, P.A.; Kenyon, J.B.; Connolly, C.; Somlo, S. Autosomal dominant polycystic kidney disease: Localization of the second gene to chromosome 4q13-q23. Genomics 1993, 18, 467–472. [Google Scholar] [CrossRef]
- Gendron, K.; Owczarek-Lipska, M.; Lang, J.; Leeb, T. Maine Coon renal screening: Ultrasonographical characterisation and preliminary genetic analysis for common genes in cats with renal cysts. J. Feline Med. Surg. 2013, 15, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Helps, C.; Tasker, S.; Harley, R. Correlation of the feline PKD1 genetic mutation with cases of PKD diagnosed by pathological examination. Exp. Mol. Pathol. 2007, 83, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Volta, A.; Manfredi, S.; Gnudi, G.; Gelati, A.; Bertoni, G. Polycystic Kidney Disease in a Chartreux Cat. J. Feline Med. Surg. 2010, 12, 138–140. [Google Scholar] [CrossRef]
- Barrs, V.R.; Gunew, M.; Foster, S.F.; Beatty, J.A.; Malik, R. Prevalence of autosomal dominant polycystic kidney disease in Persian cats and related-breeds in Sydney and Brisbane. Aust. Vet. J. 2001, 79, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Kawarasaki, M.; Aono, A.; Cho, J.; Hashimoto, T.; Sato, R. Renoprotective effects of docosahexaenoic acid in cats with early chronic kidney disease due to polycystic kidney disease: A pilot study. J. Feline Med. Surg. 2022, 24, 505–512. [Google Scholar] [CrossRef]
- Gerwing, M.; Michele, U.; Kramer, M. PKD (polycystic kidney disease)-Polyzystisches syndrome. Prakt. Tierarzt 1999, 80, 374–396. [Google Scholar]
- Lyons, L.A.; Biller, D.S.; Erdman, C.A.; Lipinski, M.J.; Young, A.E.; Roe, B.A.; Qin, B.; Grahn, R.A. Feline polycystic kidney disease mutation identified in PKD1. J. Am. Soc. Nephrol. 2004, 15, 2548–2555. [Google Scholar] [CrossRef] [PubMed]
- Domanjko-Petric, A.; Cernec, D.; Cotman, M. Polycystic kidney disease: A review and occurrence in Slovenia with comparison between ultrasound and genetic testing. J. Feline Med. Surg. 2008, 10, 115–119. [Google Scholar] [CrossRef]
- O’Leary, C.A.; Mackay, B.M.; Malik, R.; Edmondston, J.E.; Robinson, W.F.; Huxtable, C.R. Polycystic kidney disease in bull terriers: An autosomal dominant inherited disorder. Aust. Vet. J. 1999, 77, 361–366. [Google Scholar] [CrossRef]
- McAloose, D.; Casal, M.; Patterson, D.F.; Dambach, D.M. Polycystic kidney and liver disease in two related West Highland White Terrier litters. Vet. Pathol. 1998, 35, 77–81. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, C.A.; Mackay, B.M.; Taplin, R.H.; Atwell, R.B. Echocardiographic parameters in 14 healthy English Bull Terriers. Aust. Vet. J. 2003, 81, 535–542. [Google Scholar] [CrossRef]
- McKenna, S.C.; Carpenter, J.L. Polycystic disease of the kidney and liver in the Cairn Terrier. Vet. Pathol. 1980, 17, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, M.L.; Hannan, J. Abattoir survey of bovine kidney disease. Vet. Rec. 1983, 113, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.; Hebert, C.N.; Robins, B.C. Renal cysts in pigs: Prevalence and pathology in slaughtered pigs from a single herd. Vet. Rec. 1980, 106, 532–535. [Google Scholar] [CrossRef]
- Knox, R.V. Artificial insemination in pigs today. Theriogenology 2016, 85, 83–93. [Google Scholar] [CrossRef]
- Gao, Z.; Ruden, D.M.; Lu, X. PKD2 cation channel is required for directional sperm movement and male fertility. Curr. Biol. 2003, 13, 2175–2178. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef]
- Messa, P.; Alfieri, C.M.; Montanari, E.; Ferraresso, M.; Cerutti, R. ADPKD: Clinical issues before and after renal transplantation. J. Nephrol. 2016, 29, 755–763. [Google Scholar] [CrossRef]
- Porpiglia, F.; Checcucci, E.; Cillis, S.D.E.; Piramide, F.; Amparore, D.; Piana, A.; Volpi, G.; Granato, S.; Zamengo, D.; Stura, I.; et al. A prospective randomized controlled trial comparing target prostate biopsy alone approach vs. target plus standard in naïve patients with positive mpMRI. Minerva Urol. Nephrol. 2023, 75, 31–41. [Google Scholar] [CrossRef]
- Ambrosini, E.; Montanari, F.; Cristalli, C.P.; Capelli, I.; La Scola, C.; Pasini, A.; Graziano, C. Modifiers of Autosomal Dominant Polycystic Kidney Disease Severity: The Role of PKD1 Hypomorphic Alleles. Genes 2023, 14, 1230. [Google Scholar] [CrossRef] [PubMed]
- Roitbak, T.; Ward, C.J.; Harris, P.C.; Bacallao, R.; Ness, S.A.; Wandinger-Ness, A. A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol. Biol. Cell 2004, 15, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Boulter, C.; Mulroy, S.; Webb, S.; Fleming, S.; Brindle, K.; Sandford, R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl. Acad. Sci. USA 2001, 98, 12174–12179. [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.A.; Biller, D.S.; DiBartola, S.P.; Radin, M.J.; Wellman, M.L. Autosomal dominant polycystic kidney disease in Persian and Persian-cross cats. Vet. Pathol. 1997, 34, 117–126. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, C.A.; Mackay, B.M.; Taplin, R.H.; Atwell, R.B. Auscultation and echocardiographic findings in Bull Terriers with and without polycystic kidney disease. Aust. Vet. J. 2005, 83, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Barber, P.J.; Syme, H.M.; Rawlings, J.M.; Markwell, P.J. Feline hypertension: Clinical findings and response to antihypertensive treatment in 30 cases. J. Small Anim. Pract. 2001, 42, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Maggio, F.; DeFrancesco, T.C.; Atkins, C.E.; Pizzirani, S.; Gilger, B.C.; Davidson, M.G. Ocular lesions associated with systemic hypertension in cats: 69 cases (1985–1998). J. Am. Vet. Med. Assoc. 2000, 217, 695–702. [Google Scholar] [CrossRef]
- Littman, M.P. Spontaneous systemic hypertension in 24 cats. J. Vet. Intern. Med. 1994, 8, 79–86. [Google Scholar] [CrossRef]
- Kobayashi, D.L.; Peterson, M.E.; Graves, T.K.; Lesser, M.; Nichols, C.E. Hypertension in cats with chronic renal failure or hyperthyroidism. J. Vet. Intern. Med. 1990, 4, 58–62. [Google Scholar] [CrossRef]
- Beck, C.; Lavelle, R.B. Feline polycystic kidney disease in Persian and other cats: A prospective study using ultrasonography. Aust. Vet. J. 2001, 79, 181–184. [Google Scholar] [CrossRef]
- Drögemüller, M.; Klein, N.; Steffensen, R.L.; Keiner, M.; Jagannathan, V.; Leeb, T. PKD1 Nonsense Variant in a Lagotto Romagnolo Family with Polycystic Kidney Disease. Genes 2023, 14, 1210. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, C.A.; Ghoddusi, M.; Huxtable, C.R. Renal pathology of polycystic kidney disease and concurrent hereditary nephritis in Bull Terriers. Aust. Vet. J. 2002, 80, 353–361. [Google Scholar] [CrossRef]
- Webster, W.R.; Summers, P.M. Congenital polycystic kidney and liver syndrome in piglets. Aust. Vet. J. 1978, 54, 451. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le, G.E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, S.; Gallagher, A.R. Cell polarity and cystic kidney disease. Pediatr. Nephrol. 2013, 28, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, 9819. [Google Scholar] [CrossRef]
- Mekahli, D.; Parys, J.B.; Bultynck, G.; Missiaen, L.; De Smedt, H. Polycystins and cellular Ca2+ signaling. Cell. Mol. Life Sci. 2013, 70, 2697–2712. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Park, J.H. Mouse models of polycystic kidney disease induced by defects of ciliary proteins. BMB Rep. 2013, 46, 73–79. [Google Scholar] [CrossRef]
- Sussman, C.R.; Wang, X.; Chebib, F.T.; Torres, V.E. Modulation of polycystic kidney disease by G-protein coupled receptors and cyclic AMP signaling. Cell. Signal. 2020, 72, 109649. [Google Scholar] [CrossRef]
- Hansen, J.N.; Kaiser, F.; Leyendecker, P.; Stüven, B.; Krause, J.H.; Derakhshandeh, F.; Irfan, J.; Sroka, T.J.; Preval, K.M.; Desai, P.B.; et al. A cAMP signalosome in primary cilia drives gene expression and kidney cyst formation. EMBO Rep. 2022, 23, 54315. [Google Scholar] [CrossRef]
- Ye, H.; Wang, X.; Constans, M.M.; Sussman, C.R.; Chebib, F.T.; Irazabal, M.V.; Young, W.F., Jr.; Harris, P.C.; Kirschner, L.S.; Torres, V.E. The regulatory 1α subunit of protein kinase A modulates renal cystogenesis. Am. J. Physiol. Ren. Physiol. 2017, 313, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Formica, C.; Peters, D.J.M. Molecular pathways involved in injury-repair and ADPKD progression. Cell. Signal. 2020, 72, 109648. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 69, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Morleo, M.; Vieira, H.L.A.; Pennekamp, P.; Palma, A.; Bento-Lopes, L.; Omran, H.; Lopes, S.S.; Barral, D.C.; Franco, B. Crosstalk between cilia and autophagy: Implication for human diseases. Autophagy 2023, 19, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Reiterová, J.; Tesař, V. Autosomal Dominant Polycystic Kidney Disease: From Pathophysiology of Cystogenesis to Advances in the Treatment. Int. J. Mol. Sci. 2022, 23, 3317. [Google Scholar] [CrossRef]
- Benzing, T.; Simons, M.; Walz, G. Wnt signaling in polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1389–1398. [Google Scholar] [CrossRef]
- Waddell, S.H.; Yao, Y.; Olaizola, P.; Walker, A.; Jarman, E.J.; Gournopanos, K.; Gradinaru, A.; Christodoulou, E.; Gautier, P.; Boerrigter, M.M.; et al. A TGFβ-ECM-integrin signaling axis drives structural reconfiguration of the bile duct to promote polycystic liver disease. Sci. Transl. Med. 2023, 15, 5930. [Google Scholar] [CrossRef]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Consugar, M.B.; Chapman, A.B.; Torres, V.E.; Guay-Woodford, L.M.; Grantham, J.J.; Bennett, W.M.; Meyers, C.M.; Walker, D.L.; Bae, K.; et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 2143–2160. [Google Scholar] [CrossRef]
- Song, Y.; Wang, L.; Xu, M.; Lu, X.; Wang, Y.; Zhang, L. Molecular and functional characterization of porcine poly C binding protein 1 (PCBP1). BMC Vet. Res. 2024, 20, 25. [Google Scholar] [CrossRef]
- Young, A.E.; Biller, D.S.; Herrgesell, E.J.; Roberts, H.R.; Lyons, L.A. Feline polycystic kidney disease is linked to the PKD1 region. Mamm. Genome 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; O’Leary, C.A.; Kyaw-Tanner, M.; Sturm, R.A.; Duffy, D.L. A non-synonymous mutation in the canine Pkd1 gene is associated with autosomal dominant polycystic kidney disease in Bull Terriers. PLoS ONE 2011, 6, 22455. [Google Scholar] [CrossRef]
- Duarte-Chavez, R.; Stoltzfus, J.; Yellapu, V.; Martins, N.; Nanda, S.; Longo, S.; Geme, B.; Schneider, Y. Colonic diverticular disease in autosomal dominant polycystic kidney disease: Is there really an association? A nationwide analysis. Int. J. Colorectal Dis. 2021, 36, 83–91. [Google Scholar] [CrossRef]
- Vucicevic, M.; Slijepcevic, D.; Davitkov, D.; Avdalovic, V.; Aleksic-Kovacevic, S.; Stevanovic, J.; Stanimirovic, Z. First report of Polycystic kidney disease occurrence in Persian cats in Serbia. Vet. Ital. 2016, 52, 51–56. [Google Scholar] [PubMed]
- Belibi, F.A.; Edelstein, C.L. Unified Ultrasonographic Diagnostic Criteria for Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 6–8. [Google Scholar] [CrossRef]
- Biller, D.S.; DiBartola, S.P.; Eaton, K.A.; Pflueger, S.; Wellman, M.L.; Radin, M.J. Inheritance of polycystic kidney disease in Persian cats. J. Hered. 1996, 87, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gkekas, E.; Tang, T.Y.T.; Green, A.; Davidson, H.; Fraser, R.; Sayer, J.A.; Srivastava, S. Outcomes from the Northeast England cohort of autosomal dominant polycystic kidney disease (ADPKD) patients on tolvaptan. Front. Nephrol. 2022, 2, 984165. [Google Scholar] [CrossRef]
- Tamma, G.; Di Mise, A.; Ranieri, M.; Geller, A.; Tamma, R.; Zallone, A.; Valenti, G. The V2 receptor antagonist tolvaptan raises cytosolic calcium and prevents AQP2 trafficking and function: An in vitro and in vivo assessment. J. Cell Mol. Med. 2017, 21, 1767–1780. [Google Scholar] [CrossRef]
- Sans-Atxer, L.; Joly, D. Tolvaptan in the treatment of autosomal dominant polycystic kidney disease: Patient selection and special considerations. Int. J. Nephrol. Renovasc. Dis. 2018, 11, 41–51. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Atwood, D.J.; Lombardi, A.M.; Nguyen, K.N.; Toll, H.W.; Hopp, K.; Edelstein, C.L. A study of sirolimus and mTOR kinase inhibitor in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2019, 317, 187–196. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef]
- Torres, V.E. Treatment strategies and clinical trial design in ADPKD. Adv. Chronic Kidney Dis. 2010, 17, 190–204. [Google Scholar] [CrossRef]
- Amparore, D.; Pecoraro, A.; Piramide, F.; Verri, P.; Checcucci, E.; De Cillis, S.; Piana, A.; Burgio, M.; Di Dio, M.; Manfredi, M.; et al. Three-dimensional imaging reconstruction of the kidney’s anatomy for a tailored minimally invasive partial nephrectomy: A pilot study. Asian J. Urol. 2022, 9, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Capuano, I.; Buonanno, P.; Riccio, E.; Rizzo, M.; Pisani, A. Tolvaptan vs. somatostatin in the treatment of ADPKD: A review of the literature. Clin. Nephrol. 2022, 97, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kramers, B.J.; Koorevaar, I.W.; van Gastel, M.D.A.; van Goor, H.; Hallows, K.R.; Heerspink, H.L.; van Goor, H.; Hallows, K.R.; Heerspink, H.L.; Li, H.; et al. Effects of Hydrochlorothiazide and Metformin on Aquaresis and Nephroprotection by a Vasopressin V2 Receptor Antagonist in ADPKD: A Randomized Crossover Trial. Clin. J. Am. Soc. Nephrol. 2022, 17, 507–517. [Google Scholar] [CrossRef]
- Ledford, H. CRISPR gene therapy shows promise against blood diseases. Nature 2020, 588, 383. [Google Scholar] [CrossRef]
- Malech, H.L. Treatment by CRISPR-Cas9 Gene Editing—A Proof of Principle. N. Engl. J. Med. 2021, 384, 286–287. [Google Scholar] [CrossRef]
- Dodds, W.J. One Health: Animal Models of Heritable Human Bleeding Diseases. Animals 2022, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: First Regulatory Approvals for CRISPR-Cas9 Therapeutic Gene Editing for Sickle Cell Disease and Transfusion-Dependent β-Thalassemia. Med. Sci. Monit. 2024, 30, 944204. [Google Scholar] [CrossRef]
- Mehta, J. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 91. [Google Scholar]
- Geng, Y.; Zhang, L.; Fu, B.; Zhang, J.; Hong, Q.; Hu, J.; Li, D.; Luo, C.; Cui, S.; Zhu, F.; et al. Mesenchymal stem cells ameliorate rhabdomyolysis-induced acute kidney injury via the activation of M2 macrophages. Stem Cell Res. Ther. 2014, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Morigi, M.; Cerullo, D.; Introna, M.; Colpani, O.; Corna, D.; Capelli, C.; Rabelink, T.J.; Leuning, D.G.; Rottoli, D.; et al. Therapeutic potential of stromal cells of non-renal or renal origin in experimental chronic kidney disease. Stem Cell Res. Ther. 2018, 9, 220. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, N.; Xia, C.; Wang, H.; Shao, L.; Zhou, C.; Wang, J. Mesenchymal Stem Cell Therapy in Kidney Diseases: Potential and Challenges. Cell Transplant. 2023, 32, 9636897231164251. [Google Scholar] [CrossRef]
- Perico, N.; Remuzzi, G.; Griffin, M.D.; Cockwell, P.; Maxwell, A.P.; Casiraghi, F.; Rubis, N.; Peracchi, T.; Villa, A.; Todeschini, M.; et al. Safety and Preliminary Efficacy of Mesenchymal Stromal Cell (ORBCEL-M) Therapy in Diabetic Kidney Disease: A Randomized Clinical Trial (NEPHSTROM). J. Am. Soc. Nephrol. 2023, 34, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Fry, J.L., Jr.; Koch, W.E.; Jennette, J.C.; McFarland, E.; Fried, F.A.; Mandell, J. A genetically determined murine model of infantile polycystic kidney disease. J. Urol. 1985, 134, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal models for human polycystic kidney disease. Exp. Anim. 2012, 61, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of Aerobic Glycolysis Attenuates Disease Progression in Polycystic Kidney Disease. PLoS ONE 2016, 11, 0146654. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E. Vasopressin directly regulates cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 102–108. [Google Scholar] [CrossRef]
- Shibazaki, S.; Yu, Z.; Nishio, S.; Tian, X.; Thomson, R.B.; Mitobe, M.; Louvi, A.; Velazquez, H.; Ishibe, S.; Cantley, L.G.; et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008, 17, 1505–1516. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell Biol. 2017, 37, e00337-17. [Google Scholar] [CrossRef] [PubMed]
- Calvet, J.P. The Role of Calcium and Cyclic AMP in PKD. In Polycystic Kidney Disease; Li, X., Ed.; Codon Publications: Brisbane, AU, Australia, 2015. [Google Scholar]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.; Escobar-Zarate, D.; Wells, H.H.; Constans, M.M.; Thao, K.; Smith, J.M.; Sieben, C.J.; Martell, M.R.; Kline, T.L.; Irazabal, M.V.; et al. The genetic background significantly impacts the severity of kidney cystic disease in the Pkd1RC/RC mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 2021, 99, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Schena, G.; Carmosino, M.; Chiurlia, S.; Onuchic, L.; Mastropasqua, M.; Maiorano, E.; Schena, F.P.; Caplan, M.J. β3 adrenergic receptor as potential therapeutic target in ADPKD. Physiol. Rep. 2021, 9, 15058. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I.; Bhardwaj, R.; Ishikawa, Y.; Khumsubdee, S.; Krappitz, M.; Gubina, N.; Volpe, I.; Andrade, D.C.; Westergerling, P.; Staudner, T.; et al. A synthetic agent ameliorates polycystic kidney disease by promoting apoptosis of cystic cells through increased oxidative stress. Proc. Natl. Acad. Sci. USA 2024, 121, 2317344121. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Kubly, V.J.; Consugar, M.B.; Hopp, K.; Roy, S.; Horsley, S.W.; Chauveau, D.; Hopp, K.; Roy, S.; Horsley, S.W.; et al. PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009, 75, 848–855. [Google Scholar] [CrossRef]
- He, J.; Ye, J.; Li, Q.; Feng, Y.; Bai, X.; Chen, X.; Wu, C.; Yu, Z.; Zhao, Y.; Hu, X.; et al. Construction of a transgenic pig model overexpressing polycystic kidney disease 2 (PKD2) gene. Transgenic Res. 2013, 22, 861–867. [Google Scholar] [CrossRef]
- He, J.; Li, Q.; Fang, S.; Guo, Y.; Liu, T.; Ye, J.; Yu, Z.; Zhang, R.; Zhao, Y.; Hu, X.; et al. PKD1 mono-allelic knockout is sufficient to trigger renal cystogenesis in a mini-pig model. Int. J. Biol. Sci. 2015, 11, 361–369. [Google Scholar] [CrossRef]
- Watanabe, M.; Umeyama, K.; Nakano, K.; Matsunari, H.; Fukuda, T.; Matsumoto, K.; Tajiri, S.; Yamanaka, S.; Hasegawa, K.; Okamoto, K.; et al. Generation of heterozygous PKD1 mutant pigs exhibiting early-onset renal cyst formation. Lab. Investig. 2022, 102, 560–569. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Kobayashi, K.; Nakaya, M.; Iwatani, C.; Seita, Y.; Tsuchiya, H.; Matsushita, J.; Kitajima, K.; Kawamoto, I.; Nakagawa, T.; et al. Monkeys mutant for PKD1 recapitulate human autosomal dominant polycystic kidney disease. Nat. Commun. 2019, 10, 5517. [Google Scholar] [CrossRef]
- Kim, D.; Le, Q.V.; Wu, Y.; Park, J.; Oh, Y.K. Nanovesicle-Mediated Delivery Systems for CRISPR/Cas Genome Editing. Pharmaceutics 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Wu, S.; Capecchi, M.R.; Jaenisch, R. A brief review of genome editing technology for generating animal models. Front. Agric. Sci. Eng. 2020, 7, 123–128. [Google Scholar] [CrossRef]
- Nørregaard, R.; Mutsaers, H.A.M.; Frøkiær, J.; Kwon, T.H. Obstructive nephropathy and molecular pathophysiology of renal interstitial fibrosis. Physiol. Rev. 2023, 103, 2827–2872. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Matsumoto, T.; Hiratsuka, K.; Garcia Saiz, E.; Galichon, P.; Miyoshi, T.; Susa, K.; Tatsumoto, N.; Yamashita, M.; Morizane, R. Modeling injury and repair in kidney organoids reveals that homologous recombination governs tubular intrinsic repair. Sci. Transl. Med. 2022, 14, 4772. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Micropuncturing the nephron. Pflug. Arch. 2009, 458, 189–201. [Google Scholar] [CrossRef]
- Iwase, H.; Klein, E.C.; Cooper, D.K. Physiologic Aspects of Pig Kidney Transplantation in Nonhuman Primates. Comp. Med. 2018, 68, 332–340. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Human | Dog | Cat | Livestock |
|---|---|---|---|---|
| Cyst Formation | Kidneys, liver | Kidneys, liver | Kidneys, liver | Kidneys, liver |
| Additional Organ Involvement | Liver, pancreas, spleen, thyroid | Liver, pancreas, uterus, abdominalcavity | Liver, pancreas, uterus, abdominal cavity | Ascites, bile ducthyperplasia |
| Histological Features | Interstitial inflammation, fibrosis, EMT, ECM accumulatione | Proliferation of renal Tubular epithelium, heartdisease | Increased arterial pressure, proliferation ofcyst lining epithelial cells | Atrophy, degeneration, fibrous connective tissue, cyst formation |
| Clinical Progression | Chronic renal failure, hypertension, cardiovascular complications | Gradual deterioration, end-stage renal disease | Early renal failure, variecsymptoms, end-stage renal disease | Organ enlargement, reduced production efficiency |
| Species | Human | Dog | Cat | Livestock |
|---|---|---|---|---|
| Genetic Factors | Shared gene mutations, familial inheritance | Familial inheritance, breed-specific mutations | Familial inheritance, breed-specific mutations | Familial inheritance, breed-specific mutations |
| Research Approaches | Genetic sequencing, geneexpresslon analysis | Comparative genomics, histological studies | Genetic testing | Genetic mapping, histopathological analysis |
| Commonalities Identified | Pathways involving fibrosis, ECM accumulation | Renal tubular epithelial cell proliferation | Arterial pressure increase, cystlining epithelial cell proliferation | Renal and liver cyst formation, fibrous connective tissue |
| Species | Human | Dog | Cat | Livestock |
|---|---|---|---|---|
| Gene | PKD1 PKD2 | PKD1 | PKD1 | Unknown |
| Mutation | Various deletions, insertions, substitutions in PKD1 gene | G>A mutation on chromosome 29 | C.10063C>A mutation in exon 32 | Unknown mutations |
| Phenotypic Effect | Precipitation of symptom manifestation, contribution to ADPKD development | Associated with bull terrier polycystic kidney disease (BTPKD) | Premature termination codon, loss of 25% of PC-1 C-terminus | Resemble human ADPKD symptoms and pathology |
| Diagnostic Method | Human Clinical Practice | Veterinary Clinical Practice |
|---|---|---|
| Clinical Symptoms | Headaches, insomnia, blurred vision, hypertension, proteinuria, hematuria, renalpain, kidney stones, urinary tract infections, intracranial aneurysms, heart valve abnormalities | Loss of appetite, lethargy, dehydration, weight loss, increased thirst andurination, reduced urine output, anuria, palpable kidney enlargement and irregular texture |
| Imaging studies | Ultrasound, CT scanning, MRI | Ultrasound |
| Biomarkers | Blood indicators: urea, creatinine, phosphorus | Blood indicators: urea, creatinine, phosphorus |
| Genetic Testing | Identifies mutations in PKD1 and PKD2 genes | Identifies mutations in PKD1 and PKD2 genes |
| Diagnostic Criteria | Based on cyst counts on ultrasound, family history | Based on cyst counts on ultrasound, family history |
| Structure and Index | Human | Pig | Dog | Mouse |
|---|---|---|---|---|
| Kidney weight | 120–150 g | 150 g | 39 g | 0.176 g |
| Renal volume | 120–490 cm3 | 38–118 cm3 | 20–34 cm3 | 0.34 cm3 |
| Renal papilla | Multipapilla | Multipapilla | Monopapilla | Monopapilla |
| Bilateral nephron | 2–3 million | 2.2 million | 0.8 million | 30,000 |
| Glomerular filtration rate | 80–120 mL/min | 100 mL/min | 61.3 mL/min | 0.28 mL/min |
| Filtration fraction | 0.20 | 0.24 | 0.32 | 0.23 |
| Relative medullary thickness | 3.0 | 1 | 4.3 | 5.6 |
| Urine pH | 4.5–8 | 6.25–7.55 | 6–7 | 7.3–8.5 |
| 24-h urine volume | 1–2 L | 2–5 L | 0.25–1 L | 0.001–0.003 L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Zhang, Y.; Jayaprakash, S.; Zhuang, L.; He, J. Cross-Species Insights into Autosomal Dominant Polycystic Kidney Disease: Provide an Alternative View on Research Advancement. Int. J. Mol. Sci. 2024, 25, 5646. https://doi.org/10.3390/ijms25115646
Luo J, Zhang Y, Jayaprakash S, Zhuang L, He J. Cross-Species Insights into Autosomal Dominant Polycystic Kidney Disease: Provide an Alternative View on Research Advancement. International Journal of Molecular Sciences. 2024; 25(11):5646. https://doi.org/10.3390/ijms25115646
Chicago/Turabian StyleLuo, Jianing, Yuan Zhang, Sakthidasan Jayaprakash, Lenan Zhuang, and Jin He. 2024. "Cross-Species Insights into Autosomal Dominant Polycystic Kidney Disease: Provide an Alternative View on Research Advancement" International Journal of Molecular Sciences 25, no. 11: 5646. https://doi.org/10.3390/ijms25115646
APA StyleLuo, J., Zhang, Y., Jayaprakash, S., Zhuang, L., & He, J. (2024). Cross-Species Insights into Autosomal Dominant Polycystic Kidney Disease: Provide an Alternative View on Research Advancement. International Journal of Molecular Sciences, 25(11), 5646. https://doi.org/10.3390/ijms25115646