
Cloning, Expression, Purification, and Characterization of Lactate Dehydrogenase from Plasmodium knowlesi: A Zoonotic Malaria Parasite

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Homology Analysis of Amino Acid Sequence of PkLDH with Other Plasmodium spp. LDH
2.2. Construction of Recombinant Plasmid for PkLDH Expression and Purification
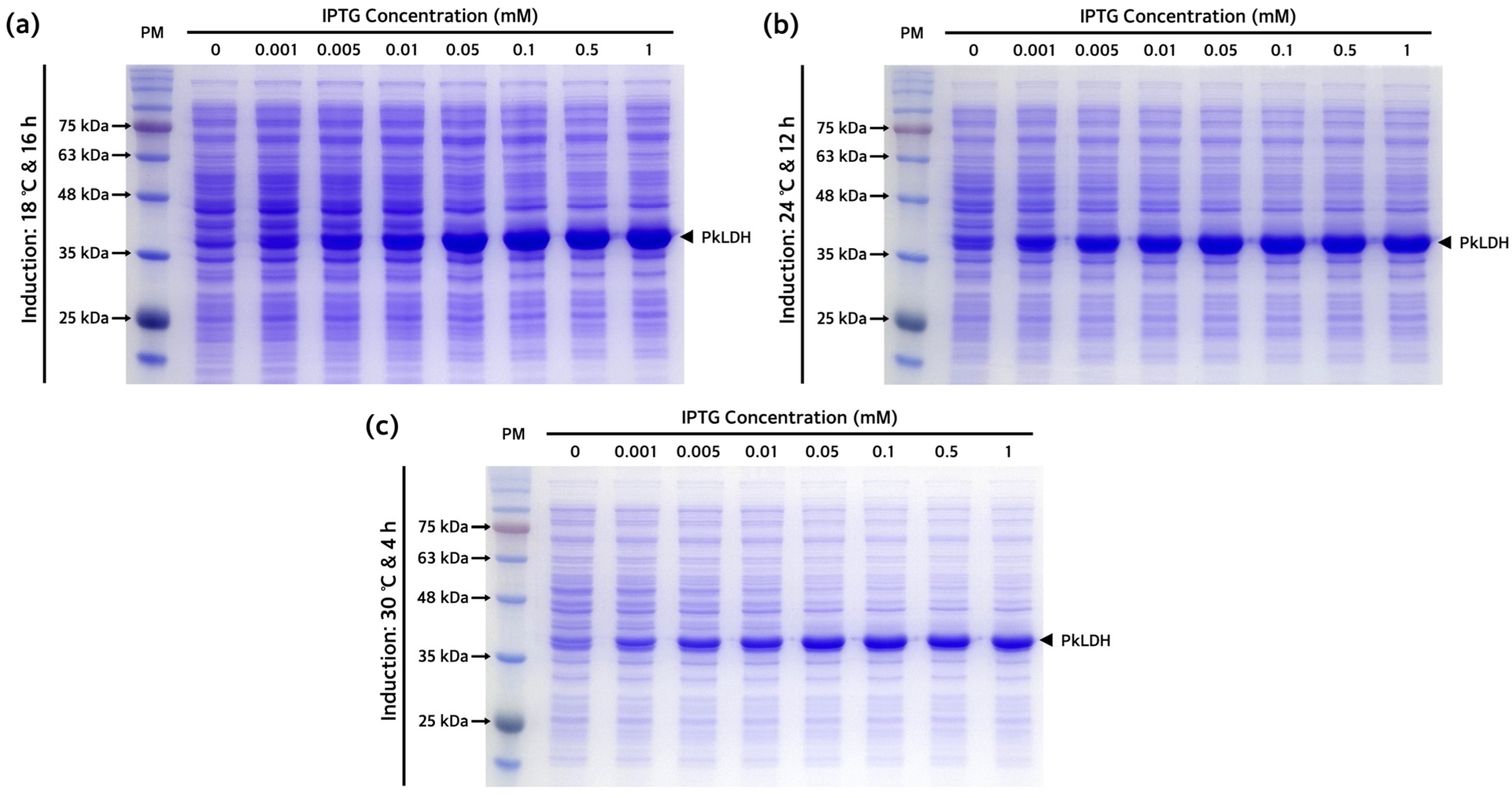
2.3. Optimization of Recombinant PkLDH Overexpression
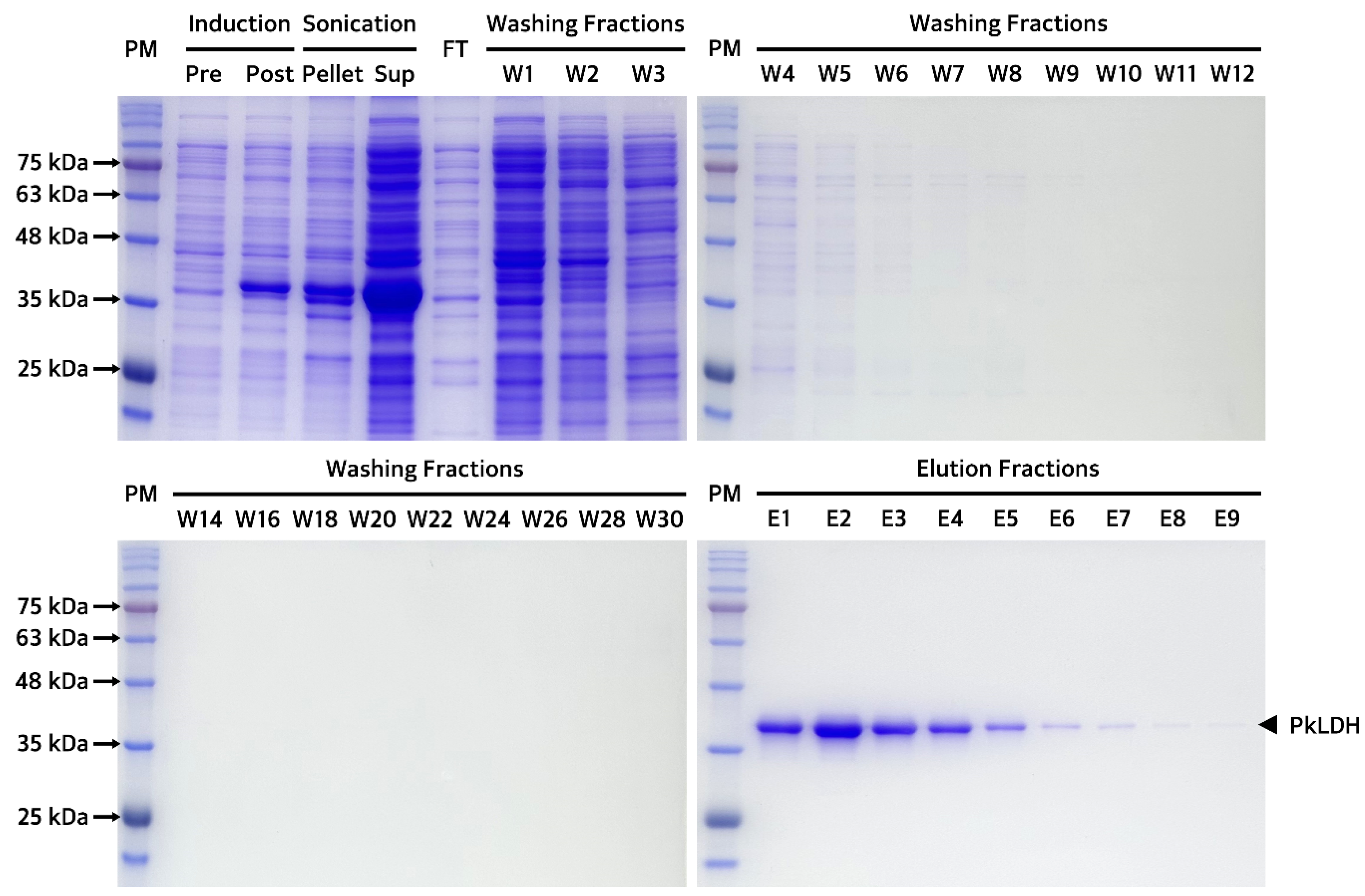
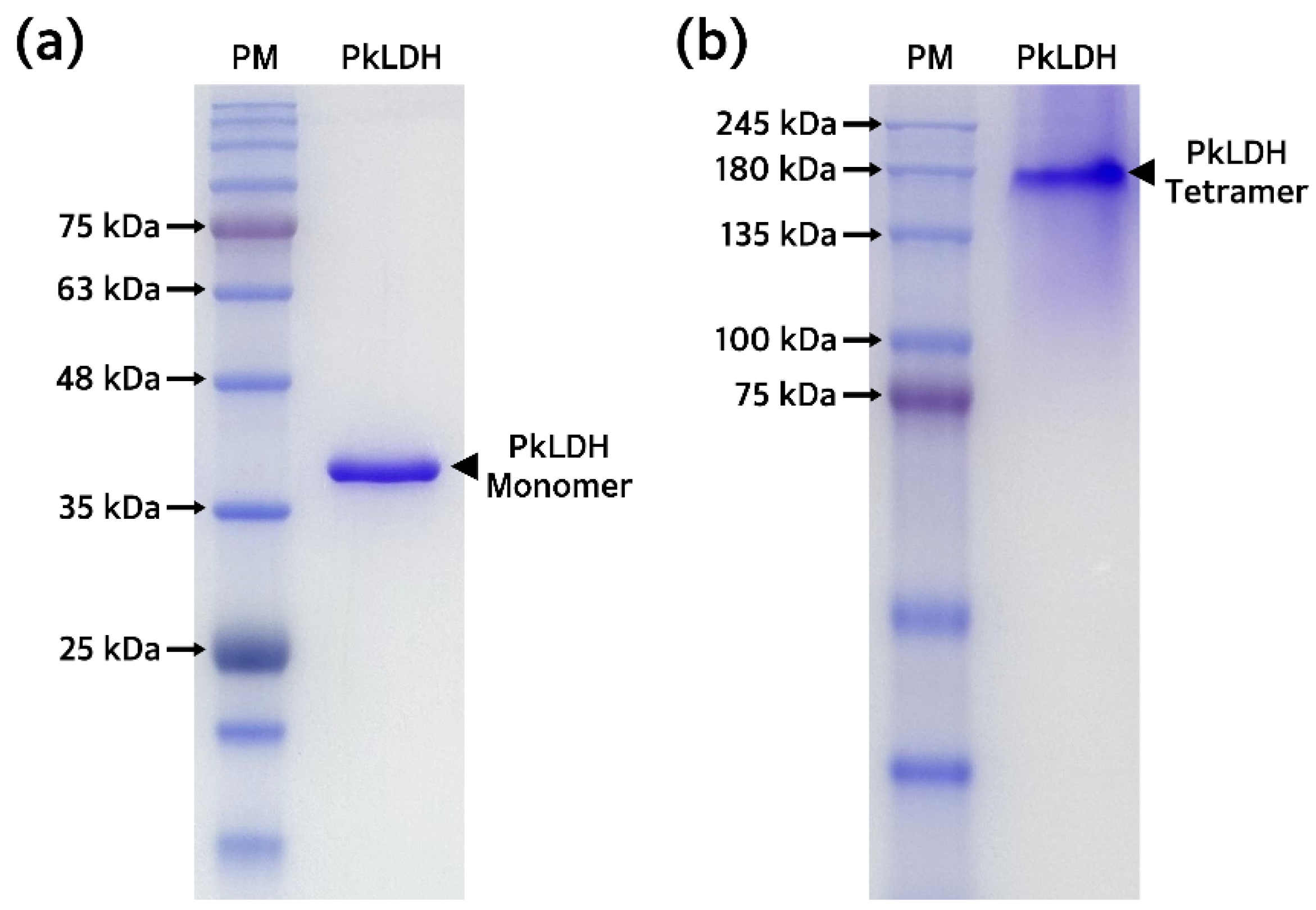
2.4. Purification and Biochemical Analysis of Recombinant PkLDH
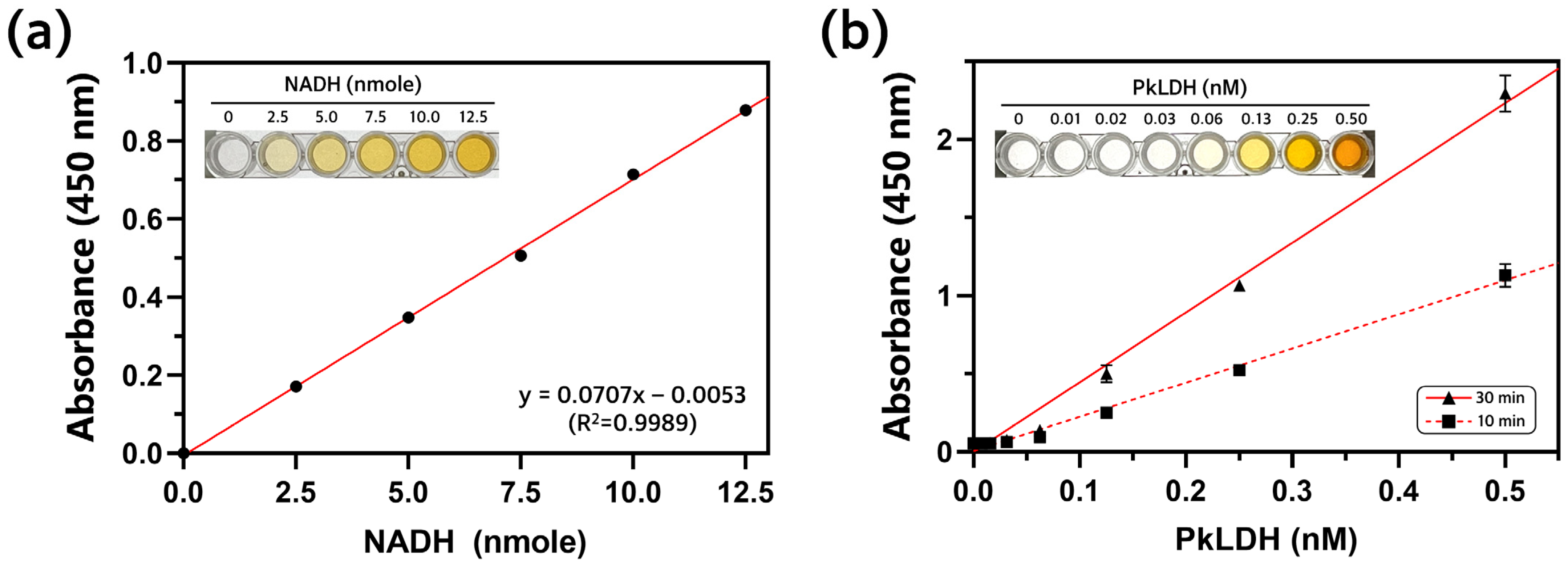
2.5. Measurement of PkLDH Activity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Gene Cloning of PkLDH and Transformation of E. coli DH5α Strain
4.3. Transformation of Rosetta(DE3) Strain and Optimization of PkLDH Expression
4.4. Isolation and Purification of Recombinant PkLDH
4.5. Biochemical Analysis of Recombinant PkLDH
4.6. Analysis of PkLDH Activity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- World Health Organization. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Ahmed, M.A.; Cox-Singh, J. Plasmodium knowlesi—An Emerging Pathogen. ISBT Sci. Ser. 2015, 10, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Lee, K.S.; Matusop, A.; Radhakrishnan, A.; Shamsul, S.S.G.; Cox-Singh, J.; Thomas, A.; Conway, D.J. A Large Focus of Naturally Acquired Plasmodium knowlesi Infections in Human Beings. Lancet 2004, 363, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- William, T.; Jelip, J.; Menon, J.; Anderios, F.; Mohammad, R.; Mohammad, T.A.A.; Grigg, M.J.; Yeo, T.W.; Anstey, N.M.; Barber, B.E. Changing Epidemiology of Malaria in Sabah, Malaysia: Increasing Incidence of Plasmodium knowlesi. Malar. J. 2014, 13, 390. [Google Scholar] [CrossRef] [PubMed]
- Kantele, A.; Jokiranta, T.S. Review of Cases with the Emerging Fifth Human Malaria Parasite, Plasmodium knowlesi. Clin. Infect. Dis. 2011, 52, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Cheong, F.W.; Amir, A.; Lai, M.Y.; Tan, J.H.; Phang, W.K.; Shahari, S.; Lau, Y.L. Plasmodium knowlesi: The Game Changer for Malaria Eradication. Malar. J. 2022, 21, 140. [Google Scholar] [CrossRef] [PubMed]
- Kotepui, M.; Kotepui, K.U.; Milanez, G.D.; Masangkay, F.R. Prevalence of Severe Plasmodium knowlesi Infection and Risk Factors Related to Severe Complications Compared with Non-severe P. knowlesi and Severe P. falciparum Malaria: A Systematic Review and Meta-analysis. Infect. Dis. Poverty 2020, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Imai, N.; White, M.T.; Ghani, A.C.; Drakeley, C.J. Transmission and Control of Plasmodium knowlesi: A Mathematical Modelling Study. PLoS Negl. Trop. Dis. 2014, 8, e2978. [Google Scholar] [CrossRef] [PubMed]
- William, T.; Rahman, H.A.; Jelip, J.; Ibrahim, M.Y.; Menon, J.; Grigg, M.J.; Yeo, T.W.; Anstey, N.M.; Barber, B.E. Increasing Incidence of Plasmodium knowlesi Malaria Following Control of P. falciparum and P. vivax Malaria in Sabah, Malaysia. PLoS Negl. Trop. Dis. 2013, 7, e2026. [Google Scholar] [CrossRef]
- Cox-Singh, J.; Davis, T.M.E.; Lee, K.S.; Shamsul, S.S.G.; Matusop, A.; Ratnam, S.; Rahman, H.A.; Conway, D.J.; Singh, B. Plasmodium knowlesi Malaria in Humans Is Widely Distributed and Potentially Life Threatening. Clin. Infect. Dis. 2008, 46, 165–171. [Google Scholar] [CrossRef]
- Amir, A.; Cheong, F.W.; de Silva, J.R.; Liew, J.W.K.; Lau, Y.L. Plasmodium knowlesi Malaria: Current Research Perspectives. Infect. Drug Resist. 2018, 11, 1145–1155. [Google Scholar] [CrossRef]
- Singh, B.; Daneshvar, C. Human Infections and Detection of Plasmodium knowlesi. Clin. Microbiol. Rev. 2013, 26, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, C.; Davis, T.M.; Cox-Singh, J.; Rafa’ee, M.Z.; Zakaria, S.K.; Divis, P.C.; Singh, B. Clinical and Parasitological Response to Oral Chloroquine and Primaquine in Uncomplicated Human Plasmodium knowlesi Infections. Malar. J. 2010, 9, 238. [Google Scholar] [CrossRef] [PubMed]
- Mahittikorn, A.; Masangkay, F.R.; Kotepui, K.U.; Milanez, G.D.J.; Kotepui, M. Quantification of the Misidentification of Plasmodium knowlesi as Plasmodium malariae by Microscopy: An Analysis of 1569 P. knowlesi Cases. Malar. J. 2021, 20, 179. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.E.; William, T.; Grigg, M.J.; Yeo, T.W.; Anstey, N.M. Limitations of Microscopy to Differentiate Plasmodium Species in A Region Co-endemic for Plasmodium falciparum, Plasmodium vivax and Plasmodium knowlesi. Malar. J. 2013, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Gitta, B.; Kilian, N. Diagnosis of Malaria Parasites Plasmodium spp. in Endemic Areas: Current Strategies for An Ancient Disease. Bioessays 2020, 42, 1900138. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.R.; O’Meara, W.P. Use of Malaria RDTs in Various Health Contexts across Sub-Saharan Africa: A Systematic Review. BMC Public Health 2017, 17, 470. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.L. Malaria Rapid Diagnostic Tests. Clin. Infect. Dis. 2012, 54, 1637–1641. [Google Scholar] [CrossRef]
- Van Hellemond, J.J.; Rutten, M.; Koelewijn, R.; Zeeman, A.M.; Verweij, J.J.; Wismans, P.J.; Kocken, C.H.; van Genderen, P.J. Human Plasmodium knowlesi Infection Detected by Rapid Diagnostic Tests for Malaria. Emerg. Infect. Dis. 2009, 15, 1478–1480. [Google Scholar] [CrossRef]
- Jain, P.; Chakma, B.; Patra, S.; Goswami, P. Potential Biomarkers and Their Applications for Rapid and Reliable Detection of Malaria. Biomed. Res. Int. 2014, 2014, 852645. [Google Scholar] [CrossRef]
- Barney, R.; Velasco, M.; Cooper, C.A.; Rashid, A.; Kyle, D.E.; Moon, R.W.; Domingo, G.J.; Jang, I.K. Diagnostic Characteristics of Lactate Dehydrogenase on A Multiplex Assay for Malaria Detection Including the Zoonotic Parasite Plasmodium knowlesi. Am. J. Trop. Med. Hyg. 2022, 106, 275–282. [Google Scholar] [CrossRef]
- Gillet, P.; Mori, M.; Esbroeck, M.V.; Ende, J.V.D.; Jacobs, J. Assessment of the Prozone Effect in Malaria Rapid Diagnostic Tests. Malar. J. 2009, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Verma, A.K.; Krishna, S.; Sharma, A.; Singh, N.; Bharti, P.K. Plasmodium falciparum Glutamate Dehydrogenase Is Genetically Conserved across Eight Malaria Endemic States of India: Exploring New Avenues of Malaria Elimination. PLoS ONE 2019, 14, e0218210. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.E.; William, T.; Grigg, M.J.; Piera, K.; Yeo, T.W.; Anstey, N.M. Evaluation of the Sensitivity of a pLDH-based and an Aldolase-based Rapid Diagnostic Test for Diagnosis of Uncomplicated and Severe Malaria Caused by PCR-confirmed Plasmodium knowlesi, Plasmodium falciparum, and Plasmodium vivax. J. Clin. Microbiol. 2013, 51, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Fuad, F.A.A.; Salim, N.O. Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays. Processes 2022, 10, 2443. [Google Scholar] [CrossRef]
- Fokou, P.V.T.; Tali, B.M.T.; Dize, D.; Mbouna, C.D.J.; Ngansop, C.A.N.; Keumoe, R.; Tchokouaha, L.R.Y.; Tchouankeu, J.C.; Escudie, F.; Duffy, J.; et al. Implementation and Continued Validation of the Malaria Plasmodium falciparum Lactate Dehydrogenase-based Colorimetric Assay for Use in Antiplasmodial Drug Screening. Anal. Biochem. 2022, 648, 114669. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.F.; binti Sakam, S.S.; Rajahram, G.S.; William, T.; Isnadi, M.F.A.R.; Daim, S.; Barber, B.E.; Kho, S.; Sutherland, C.J.; Anstey, N.M.; et al. Diagnostic Accuracy and Limit of Detection of Ten Malaria Parasite Lactate Dehydrogenase-based Rapid Tests for Plasmodium knowlesi and P. falciparum. Front. Cell. Infect. Microbiol. 2022, 12, 1023219. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, J.W. Enzyme-linked Aptamer-based Sandwich Assay (ELASA) for Detecting Plasmodium falciparum Lactate Dehydrogenase, A Malarial Biomarker. RSC Adv. 2022, 12, 29535–29542. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Pouresmaeil, M.; Azizi-Dargahlou, S. Factors Involved in Heterologous Expression of Proteins in E. coli Host. Arch. Microbiol. 2023, 205, 212. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Nong, F.T.; Wang, Y.Z.; Yan, C.X.; Gu, Y.; Song, P.; Sun, X.M. Strategies for Efficient Production of Recombinant Proteins in Escherichia coli: Alleviating the Host Burden and Enhancing Protein Activity. Microb. Cell Fact. 2022, 21, 191. [Google Scholar] [CrossRef]
- Jew, K.; Smith, P.E.J.; So, B.; Kasman, J.; Oza, J.P.; Black, M.W. Characterizing and Improving pET Vectors for Cell-free Expression. Front. Bioeng. Biotechnol. 2022, 10, 895069. [Google Scholar] [CrossRef] [PubMed]
- Shilling, P.J.; Mirzadeh, K.; Cumming, A.J.; Widesheim, M.; Köck, Z.; Daley, D.O. Improved Designs for pET Expression Plasmids Increase Protein Production Yield in Escherichia coli. Commun. Biol. 2020, 3, 214. [Google Scholar] [CrossRef]
- Choi, J.W.; Eom, H.J.; Kim, H.Y. Non-structural Protein 1 from Japanese Encephalitis Virus Expressed in E. coli Retains Its Molecular Weight and Immunogenicity. Protein Expr. Purif. 2020, 169, 105548. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Shin, J.S.; Lee, K.W.; Eom, H.J.; Jo, B.G.; Lee, J.W.; Kim, J.H.; Kim, S.Y.; Kang, J.H.; Choi, J.W. Expression, Purification, and Characterization of Plasmodium vivax Lactate Dehydrogenase from Bacteria without Codon Optimization. Int. J. Mol. Sci. 2023, 24, 11083. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Ha, S.O.; Kim, Y.J.; Shin, J.S.; Choi, M.J.; Yu, S.E.; Han, J.; Park, E.J.; Park, K.S.; Kang, J.H. Characterization of Escherichia coli Strains for Novel Production of Plasmodium ovale Lactate Dehydrogenase. Microorganisms 2024, 12, 876. [Google Scholar] [CrossRef]
- San-Miguel, T.; Pérez-Bermúdez, P.; Gavidia, I. Production of Soluble Eukaryotic Recombinant Proteins in E. coli Is Favoured in Early Log-phase Cultures Induced at Low Temperature. SpringerPlus 2013, 2, 89. [Google Scholar] [CrossRef]
- Francis, D.M.; Page, R. Strategies to Optimize Protein Expression in E. coli. Curr. Protoc. Protein Sci. 2010, 61, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.P.; Mortensen, K.K. Soluble Expression of Recombinant Proteins in the Cytoplasm of Escherichia coli. Microb. Cell Fact. 2005, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Kaushal, D.C.; Rathaur, S.; Kumar, N.; Kaushal, N.A. Cloning, Overexpression, Purification and Characterization of Plasmodium knowlesi Lactate Dehydrogenase. Protein Expr. Purif. 2012, 84, 195–203. [Google Scholar] [CrossRef]
- Salim, N.O.; Fuad, F.A.A.; Khairuddin, F.; Seman, W.M.K.W.; Jonet, M.A. Purifying and Characterizing Bacterially Expressed Soluble Lactate Dehydrogenase from Plasmodium knowlesi for the Development of Anti-malarial Drugs. Molecules 2021, 26, 6625. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-W.; Choi, M.-J.; Kim, Y.-J.; Kim, S.Y. Cloning, Expression, Purification, and Characterization of Lactate Dehydrogenase from Plasmodium knowlesi: A Zoonotic Malaria Parasite. Int. J. Mol. Sci. 2024, 25, 5615. https://doi.org/10.3390/ijms25115615
Choi J-W, Choi M-J, Kim Y-J, Kim SY. Cloning, Expression, Purification, and Characterization of Lactate Dehydrogenase from Plasmodium knowlesi: A Zoonotic Malaria Parasite. International Journal of Molecular Sciences. 2024; 25(11):5615. https://doi.org/10.3390/ijms25115615
Chicago/Turabian StyleChoi, Jae-Won, Min-Ji Choi, Yeon-Jun Kim, and So Yeon Kim. 2024. "Cloning, Expression, Purification, and Characterization of Lactate Dehydrogenase from Plasmodium knowlesi: A Zoonotic Malaria Parasite" International Journal of Molecular Sciences 25, no. 11: 5615. https://doi.org/10.3390/ijms25115615
APA StyleChoi, J.-W., Choi, M.-J., Kim, Y.-J., & Kim, S. Y. (2024). Cloning, Expression, Purification, and Characterization of Lactate Dehydrogenase from Plasmodium knowlesi: A Zoonotic Malaria Parasite. International Journal of Molecular Sciences, 25(11), 5615. https://doi.org/10.3390/ijms25115615