

The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
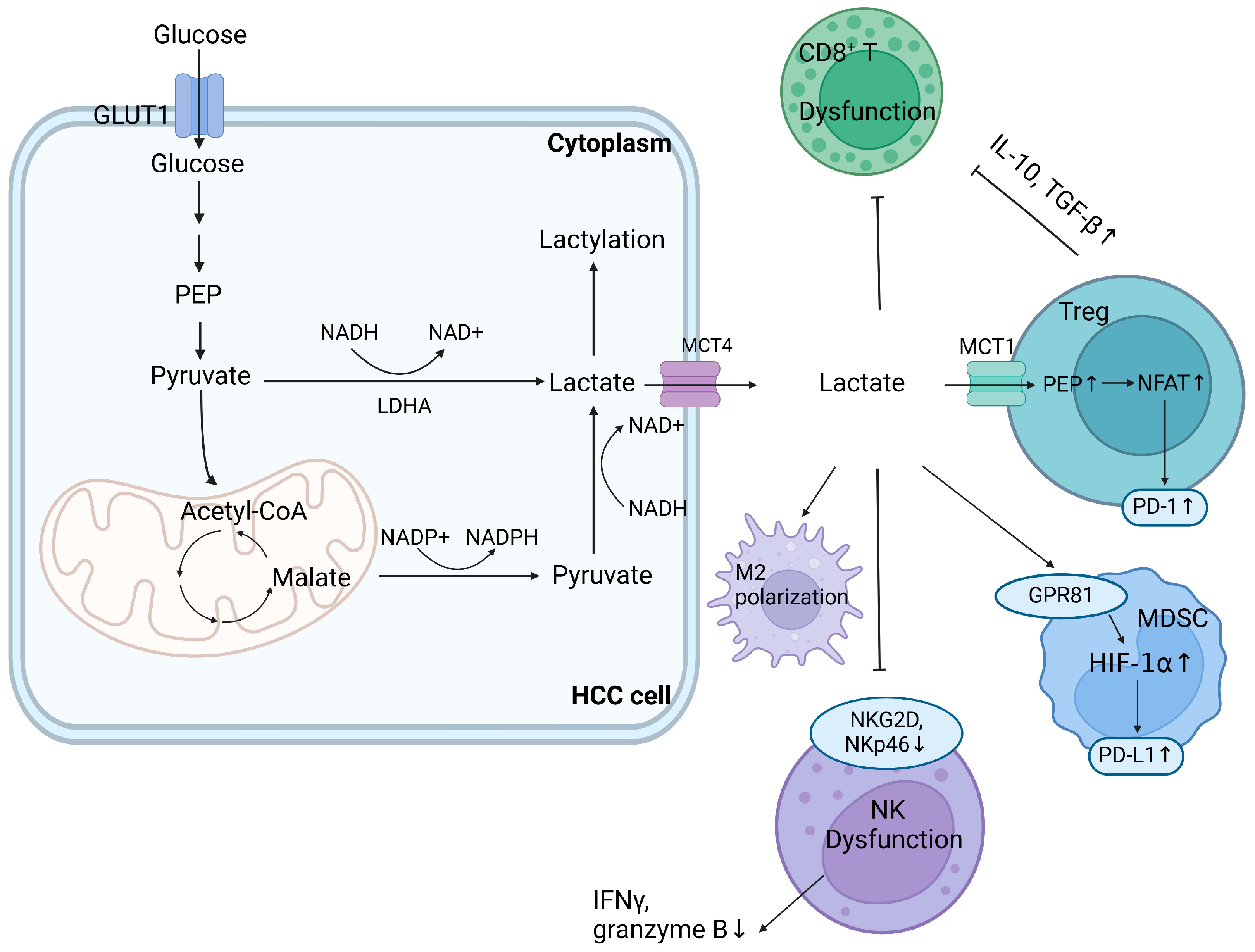
2. Impact of Dysregulated Glucose Metabolism in the TME
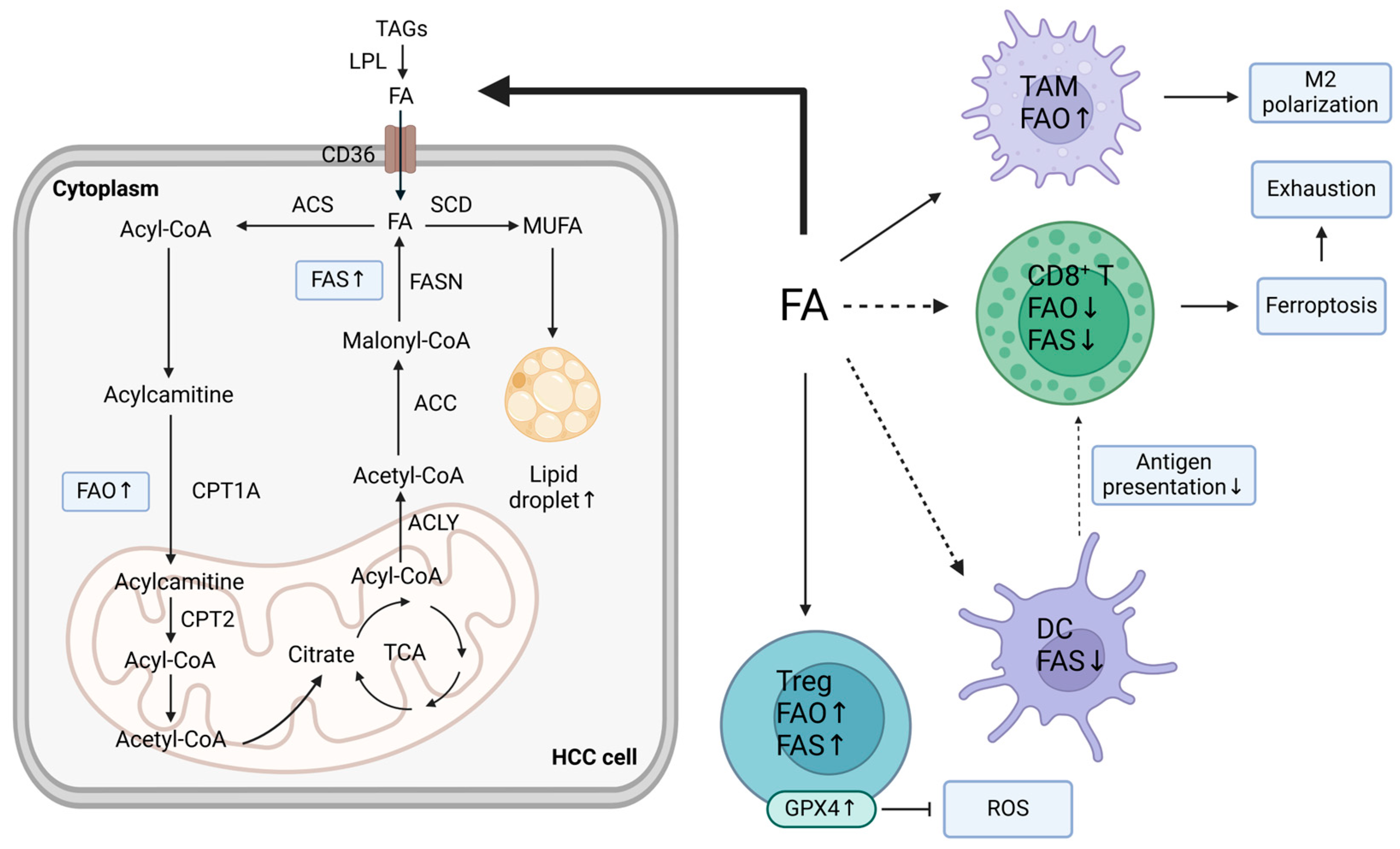
3. Impact of Dysregulated Lipid Metabolism in the TME
4. Impact of Dysregulated Amino Acid Metabolism in the TME
5. Local Metabolic Regulation
6. Targeting Metabolism to Restore Immunity
6.1. Targeting Glucose Metabolism
6.2. Targeting Lipid Metabolism
6.3. Targeting Amino Acid Metabolism
6.4. Targeting Systemic Metabolism
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- Adebayo Michael, A.O.; Ko, S.; Tao, J.; Moghe, A.; Yang, H.; Xu, M.; Russell, J.O.; Pradhan-Sundd, T.; Liu, S.; Singh, S.; et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by β-Catenin Mutations. Cell Metab. 2019, 29, 1135–1150.e6. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Liu, Z.; Fan, J.; Liu, S.; Tan, S.; Li, X.; Li, B.; Yang, X. Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma. Cells 2022, 11, 3924. [Google Scholar] [CrossRef]
- Corrado, M.; Edwards-Hicks, J.; Villa, M.; Flachsmann, L.J.; Sanin, D.E.; Jacobs, M.; Baixauli, F.; Stanczak, M.; Anderson, E.; Azuma, M.; et al. Dynamic Cardiolipin Synthesis Is Required for CD8(+) T Cell Immunity. Cell Metab. 2020, 32, 981–995.e7. [Google Scholar] [CrossRef]
- Mossmann, D.; Müller, C.; Park, S.; Ryback, B.; Colombi, M.; Ritter, N.; Weißenberger, D.; Dazert, E.; Coto-Llerena, M.; Nuciforo, S.; et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell 2023, 186, 5068–5083.e23. [Google Scholar] [CrossRef]
- Prasad, Y.R.; Anakha, J.; Pande, A.H. Treating liver cancer through arginine depletion. Drug Discov. Today 2024, 29, 103940. [Google Scholar] [CrossRef]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015, 4, e954829. [Google Scholar] [CrossRef]
- Waldron, T.J.; Quatromoni, J.G.; Karakasheva, T.A.; Singhal, S.; Rustgi, A.K. Myeloid derived suppressor cells: Targets for therapy. Oncoimmunology 2013, 2, e24117. [Google Scholar] [CrossRef]
- Pu, W.; Zhang, H.; Huang, X.; Tian, X.; He, L.; Wang, Y.; Zhang, L.; Liu, Q.; Li, Y.; Li, Y.; et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat. Commun. 2016, 7, 13369. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’Sullivan, J.; Sheahan, K.; Cantley, L.C.; Vander Heiden, M.G.; Vitkup, D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013, 31, 522–529. [Google Scholar] [CrossRef]
- Golonka, R.M.; Vijay-Kumar, M. Atypical immunometabolism and metabolic reprogramming in liver cancer: Deciphering the role of gut microbiome. Adv. Cancer Res. 2021, 149, 171–255. [Google Scholar]
- Benhamouche, S.; Decaens, T.; Godard, C.; Chambrey, R.; Rickman, D.S.; Moinard, C.; Vasseur-Cognet, M.; Kuo, C.J.; Kahn, A.; Perret, C.; et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell. 2006, 10, 759–770. [Google Scholar] [CrossRef]
- Braeuning, A.; Ittrich, C.; Köhle, C.; Hailfinger, S.; Bonin, M.; Buchmann, A.; Schwarz, M. Differential gene expression in periportal and perivenous mouse hepatocytes. FEBS J. 2006, 273, 5051–5061. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Wuensch, T.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhütter, H.G. Metabolic heterogeneity of human hepatocellular carcinoma: Implications for personalized pharmacological treatment. FEBS J. 2021, 288, 2332–2346. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Zeng, Z.; Lan, J.; Lei, S.; Yang, Y.; He, Z.; Xue, Y.; Chen, T. Simultaneous Inhibition of Ornithine Decarboxylase 1 and Pyruvate Kinase M2 Exerts Synergistic Effects Against Hepatocellular Carcinoma Cells. Onco Targets Ther. 2020, 13, 11697–11709. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, P.; Deng, K. MYC Promotes LDHA Expression through MicroRNA-122-5p to Potentiate Glycolysis in Hepatocellular Carcinoma. Anal. Cell. Pathol. 2022, 2022, 1435173. [Google Scholar] [CrossRef]
- Fiume, L.; Vettraino, M.; Manerba, M.; Di Stefano, G. Inhibition of lactic dehydrogenase as a way to increase the anti-proliferative effect of multi-targeted kinase inhibitors. Pharmacol. Res. 2011, 63, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Daneshmandi, S.; Choi, J.E.; Yan, Q.; MacDonald, C.R.; Pandey, M.; Goruganthu, M.; Roberts, N.; Singh, P.K.; Higashi, R.M.; Lane, A.N.; et al. Myeloid-derived suppressor cell mitochondrial fitness governs chemotherapeutic efficacy in hematologic malignancies. Nat. Commun. 2024, 15, 2803. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Huang, C.; Li, N.; Zou, L.; Chia, S.E.; Chen, S.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Comparison of hepatic and serum lipid signatures in hepatocellular carcinoma patients leads to the discovery of diagnostic and prognostic biomarkers. Oncotarget 2018, 9, 5032–5043. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Bu, L.; Zhang, Z.; Chen, J.; Fan, Y.; Guo, J.; Su, Y.; Wang, H.; Zhang, X.; Wu, X.; Jiang, Q.; et al. High-fat diet promotes liver tumorigenesis via palmitoylation and activation of AKT. Gut 2024. [Google Scholar] [CrossRef]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M.G.; Wang, J.; et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020, 69, 177–186. [Google Scholar] [CrossRef]
- Zhang, Q.; Lou, Y.; Bai, X.L.; Liang, T.B. Immunometabolism: A novel perspective of liver cancer microenvironment and its influence on tumor progression. World J. Gastroenterol. 2018, 24, 3500–3512. [Google Scholar] [CrossRef]
- Huang, F.; Wang, K.; Shen, J. Lipoprotein-associated phospholipase A2: The story continues. Med. Res. Rev. 2020, 40, 79–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, W.; Meng, F.; Jiang, Q.; Tang, W.; Liu, Z.; Lin, X.; Xue, R.; Zhang, S.; Dong, L. Inhibiting PLA2G7 reverses the immunosuppressive function of intratumoral macrophages and augments immunotherapy response in hepatocellular carcinoma. J. Immunother. Cancer 2024, 12, e008094. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Li, T.; Zheng, X.; Lin, L.; Wang, X.; Li, B.; Huang, J.; Wang, Y.; Shuai, X.; Zhu, K. Nanodrug enhances post-ablation immunotherapy of hepatocellular carcinoma via promoting dendritic cell maturation and antigen presentation. Bioact. Mater. 2023, 21, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, X.; Li, Y.; Wei, S.; He, G.; Liu, J.; Li, X.; Yang, S.; Li, D.; Lin, W.; et al. Glutamine addiction in tumor cell: Oncogene regulation and clinical treatment. Cell Commun. Signal. 2024, 22, 12. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Cyriac, R.; Lee, K. Glutaminase inhibition as potential cancer therapeutics: Current status and future applications. J. Enzyme Inhib. Med. Chem. 2024, 39, 2290911. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Zhang, Y.; Chung, S.F.; Tam, S.Y.; Leung, Y.C.; Guan, X. Arginine deprivation as a strategy for cancer therapy: An insight into drug design and drug combination. Cancer Lett. 2021, 502, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Nijhuis, A.; Keun, H.C. RNA-binding motif protein 39 (RBM39): An emerging cancer target. Br. J. Pharmacol. 2022, 179, 2795–2812. [Google Scholar] [CrossRef]
- Yu, S.J.; Ma, C.; Heinrich, B.; Brown, Z.J.; Sandhu, M.; Zhang, Q.; Fu, Q.; Agdashian, D.; Rosato, U.; Korangy, F.; et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J. Hepatol. 2019, 70, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Q.; Zhou, J.; Xiao, Z.; Liu, J.; Deng, S.; Hong, X.; Huang, W.; Cai, M.; Guo, Y.; et al. T cell-mediated targeted delivery of tadalafil regulates immunosuppression and polyamine metabolism to overcome immune checkpoint blockade resistance in hepatocellular carcinoma. J. Immunother. Cancer 2023, 11, e006493. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T. Impact of dietary carbohydrate restriction on the pathobiology of Hepatocellular Carcinoma: The gut-liver axis and beyond. Semin. Immunol. 2023, 66, 101736. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Vander Heiden, M.G. A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer 2019, 19, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Long, J.; Yang, X.; Lin, J.; Song, Y.; Xie, F.; Xun, Z.; Wang, Y.; Wang, Y.; et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J. Immunother. Cancer 2021, 9, e003334. [Google Scholar] [CrossRef]
- Lee, P.C.; Wu, C.J.; Hung, Y.W.; Lee, C.J.; Chi, C.T.; Lee, I.C.; Yu-Lun, K.; Chou, S.H.; Luo, J.C.; Hou, M.C.; et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e004779. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Ding, Y.; Qin, Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front. Immunol. 2023, 14, 1133308. [Google Scholar] [CrossRef]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef] [PubMed]
- Savic, L.J.; Chapiro, J.; Duwe, G.; Geschwind, J.F. Targeting glucose metabolism in cancer: New class of agents for loco-regional and systemic therapy of liver cancer and beyond? Hepat Oncol. 2016, 3, 19–28. [Google Scholar] [CrossRef]
- Tsai, P.C.; Kuo, H.T.; Hung, C.H.; Tseng, K.C.; Lai, H.C.; Peng, C.Y.; Wang, J.H.; Chen, J.J.; Lee, P.L.; Chien, R.N.; et al. Metformin reduces hepatocellular carcinoma incidence after successful antiviral therapy in patients with diabetes and chronic hepatitis C in Taiwan. J. Hepatol. 2023, 78, 281–292. [Google Scholar] [CrossRef]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, H.; Zhang, L.; Zhu, A.X.; Bernards, R.; Qin, W.; Wang, C. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 203–222. [Google Scholar] [CrossRef]
- Brandi, N.; Renzulli, M. The Synergistic Effect of Interventional Locoregional Treatments and Immunotherapy for the Treatment of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 8598. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Finn, R.S.; Sangro, B. Combination immunotherapy for hepatocellular carcinoma. J. Hepatol. 2023, 79, 506–515. [Google Scholar] [CrossRef]
- EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [CrossRef] [PubMed]
- Hu, B.; Yu, M.; Ma, X.; Sun, J.; Liu, C.; Wang, C.; Wu, S.; Fu, P.; Yang, Z.; He, Y.; et al. IFNα Potentiates Anti-PD-1 Efficacy by Remodeling Glucose Metabolism in the Hepatocellular Carcinoma Microenvironment. Cancer Discov. 2022, 12, 1718–1741. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, W.; Tang, Z.; Ji, X.; Zhou, Y.; Song, S.; Tian, M.; Tao, C.; Huang, R.; Zhu, G.; et al. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology 2023, 77, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro Fernández, J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. [Google Scholar] [CrossRef] [PubMed]
- van Son, K.C.; Verschuren, L.; Hanemaaijer, R.; Reeves, H.; Takkenberg, R.B.; Drenth, J.P.H.; Tushuizen, M.E.; Holleboom, A.G. Non-Parenchymal Cells and the Extracellular Matrix in Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease. Cancers 2023, 15, 1308. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Xu, Y.; Hao, X.; Ren, Y.; Xu, Q.; Liu, X.; Song, S.; Wang, Y. Research progress of abnormal lactate metabolism and lactate modification in immunotherapy of hepatocellular carcinoma. Front. Oncol. 2022, 12, 1063423. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022, 40, 201–218.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, X.; Yang, X.; Li, W.; Li, S.; Hu, Z.; Ling, C.; Shi, R.; Liu, J.; Chen, G.; et al. Dual Blockade of Lactate/GPR81 and PD-1/PD-L1 Pathways Enhances the Anti-Tumor Effects of Metformin. Biomolecules 2021, 11, 1373. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.G.; Zhou, Z.L.; Wang, X.Y.; Liu, B.Y.; Lu, J.Y.; Liu, S.; Zhang, G.B.; Zhan, M.X.; Chen, Y. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut 2022, 71, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Zhang, L.; Wang, M.; Zhou, Y.; Jia, K.; Wang, S.; Wang, M.; Li, Y.; Luo, S.; et al. Mitochondrial IRG1 traps MCL-1 to induce hepatocyte apoptosis and promote carcinogenesis. Cell Death Dis. 2023, 14, 625. [Google Scholar] [CrossRef]
- Gu, X.; Wei, H.; Suo, C.; Shen, S.; Zhu, C.; Chen, L.; Yan, K.; Li, Z.; Bian, Z.; Zhang, P.; et al. Itaconate promotes hepatocellular carcinoma progression by epigenetic induction of CD8(+) T-cell exhaustion. Nat. Commun. 2023, 14, 8154. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Z.; Liu, G.; Zhang, Y.; Liu, S.; Gan, D.; Chang, W.; Peng, X.; Sung, E.S.; Gilbert, K.; et al. Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab. 2023, 35, 1688–1703.e10. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647. [Google Scholar] [CrossRef]
- Liu, M.X.; Jin, L.; Sun, S.J.; Liu, P.; Feng, X.; Cheng, Z.L.; Liu, W.R.; Guan, K.L.; Shi, Y.H.; Yuan, H.X.; et al. Metabolic reprogramming by PCK1 promotes TCA cataplerosis, oxidative stress and apoptosis in liver cancer cells and suppresses hepatocellular carcinoma. Oncogene 2018, 37, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Liu, Z.; You, Y.; Chen, Q.; Li, G.; Pan, W.; Yang, Q.; Dong, J.; Wu, Y.; Bei, J.X.; Pan, C.; et al. Extracellular vesicle-mediated communication between hepatocytes and natural killer cells promotes hepatocellular tumorigenesis. Mol. Ther. 2022, 30, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell. 2017, 32, 807–823.e12. [Google Scholar] [CrossRef]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: Structure, function, regulation, and role in disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Y.; Zhou, H.; Xu, Y.; Cao, Y.; Cheng, C.; Peng, J.; Li, H.; Zhang, L.; Su, K.; et al. Multiomics identifies metabolic subtypes based on fatty acid degradation allocating personalized treatment in hepatocellular carcinoma. Hepatology 2024, 79, 289–306. [Google Scholar] [CrossRef]
- Murai, H.; Kodama, T.; Maesaka, K.; Tange, S.; Motooka, D.; Suzuki, Y.; Shigematsu, Y.; Inamura, K.; Mise, Y.; Saiura, A.; et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology 2023, 77, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.M.; Menk, A.V.; Shi, J.; Tsung, A.; Delgoffe, G.M.; Butterfield, L.H. Tumor-Derived α-Fetoprotein Suppresses Fatty Acid Metabolism and Oxidative Phosphorylation in Dendritic Cells. Cancer Immunol. Res. 2019, 7, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Furihata, T.; Sawada, T.; Kita, J.; Iso, Y.; Kato, M.; Rokkaku, K.; Shimoda, M.; Kubota, K. Serum alpha-fetoprotein level per tumor volume reflects prognosis in patients with hepatocellular carcinoma after curative hepatectomy. Hepatogastroenterology 2008, 55, 1705–1709. [Google Scholar] [PubMed]
- Lu, X.; Zhang, X.; Zhang, Y.; Zhang, K.; Zhan, C.; Shi, X.; Li, Y.; Zhao, J.; Bai, Y.; Wang, Y.; et al. Metabolic profiling analysis upon acylcarnitines in tissues of hepatocellular carcinoma revealed the inhibited carnitine shuttle system caused by the downregulated carnitine palmitoyltransferase 2. Mol. Carcinog. 2019, 58, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lin, J.Z.; Yang, X.B.; Sang, X.T. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Prolif. 2020, 53, e12772. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Q.; Tang, Z.; Huang, R.; Qu, W.F.; Fang, Y.; Yang, R.; Tao, C.Y.; Gao, J.; Wu, X.L.; Sun, H.X.; et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023, 9, 25. [Google Scholar] [CrossRef]
- Huang, H.; Tsui, Y.M.; Ng, I.O. Fueling HCC Dynamics: Interplay Between Tumor Microenvironment and Tumor Initiating Cells. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 1105–1116. [Google Scholar] [CrossRef]
- Akkız, H. Emerging Role of Cancer-Associated Fibroblasts in Progression and Treatment of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3941. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, H.; Cheng, S.; Chen, Y.; He, L.; Ren, J.; He, X.; Chen, J.; Zheng, L.; Li, F. Glutamate dehydrogenase 1 mediated glutaminolysis sustains HCC cells survival under glucose deprivation. J. Cancer 2022, 13, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Tang, X.; Ren, Y.; Yang, Y.; Song, F.; Fu, J.; Liu, S.; Yu, M.; Chen, J.; Wang, S.; et al. An RNA-RNA crosstalk network involving HMGB1 and RICTOR facilitates hepatocellular carcinoma tumorigenesis by promoting glutamine metabolism and impedes immunotherapy by PD-L1+ exosomes activity. Signal Transduct. Target. Ther. 2021, 6, 421. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Lévy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, N.; Li, H.; Tao, C.; Xiao, T.; Rong, W. The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2024, 25, 5584. https://doi.org/10.3390/ijms25115584
Hu N, Li H, Tao C, Xiao T, Rong W. The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2024; 25(11):5584. https://doi.org/10.3390/ijms25115584
Chicago/Turabian StyleHu, Nan, Haiyang Li, Changcheng Tao, Ting Xiao, and Weiqi Rong. 2024. "The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma" International Journal of Molecular Sciences 25, no. 11: 5584. https://doi.org/10.3390/ijms25115584
APA StyleHu, N., Li, H., Tao, C., Xiao, T., & Rong, W. (2024). The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 25(11), 5584. https://doi.org/10.3390/ijms25115584