
Genome-Wide Identification and Characterization of Homeobox Transcription Factors in Phoma sorghina var. saccharum Causing Sugarcane Twisted Leaf Disease
 ,
,  and
and
Abstract
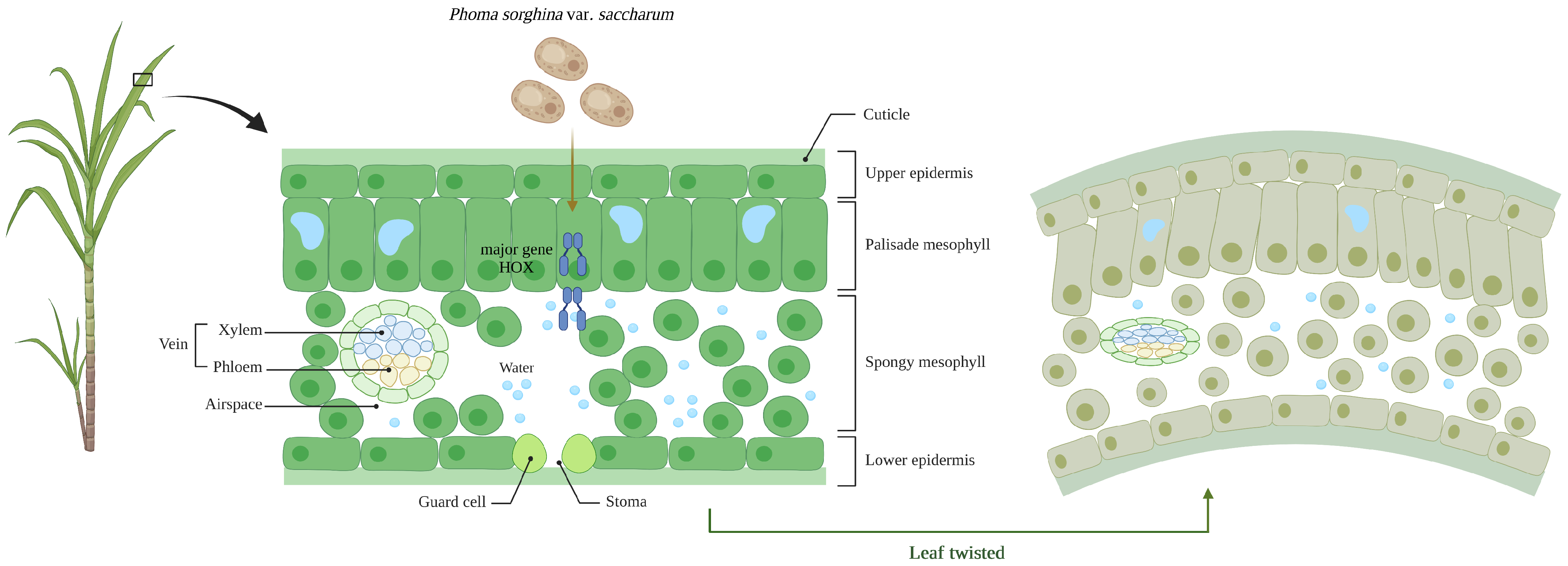
1. Introduction
2. Results
2.1. Assessment of Genome Assembly and Completeness
2.2. Gene Annotation
2.3. Identification of PsHOX Gene Family
2.4. Characterization of PsHOX Gene Family and Evolutionary Analysis
2.5. Verification of Knockout Mutants
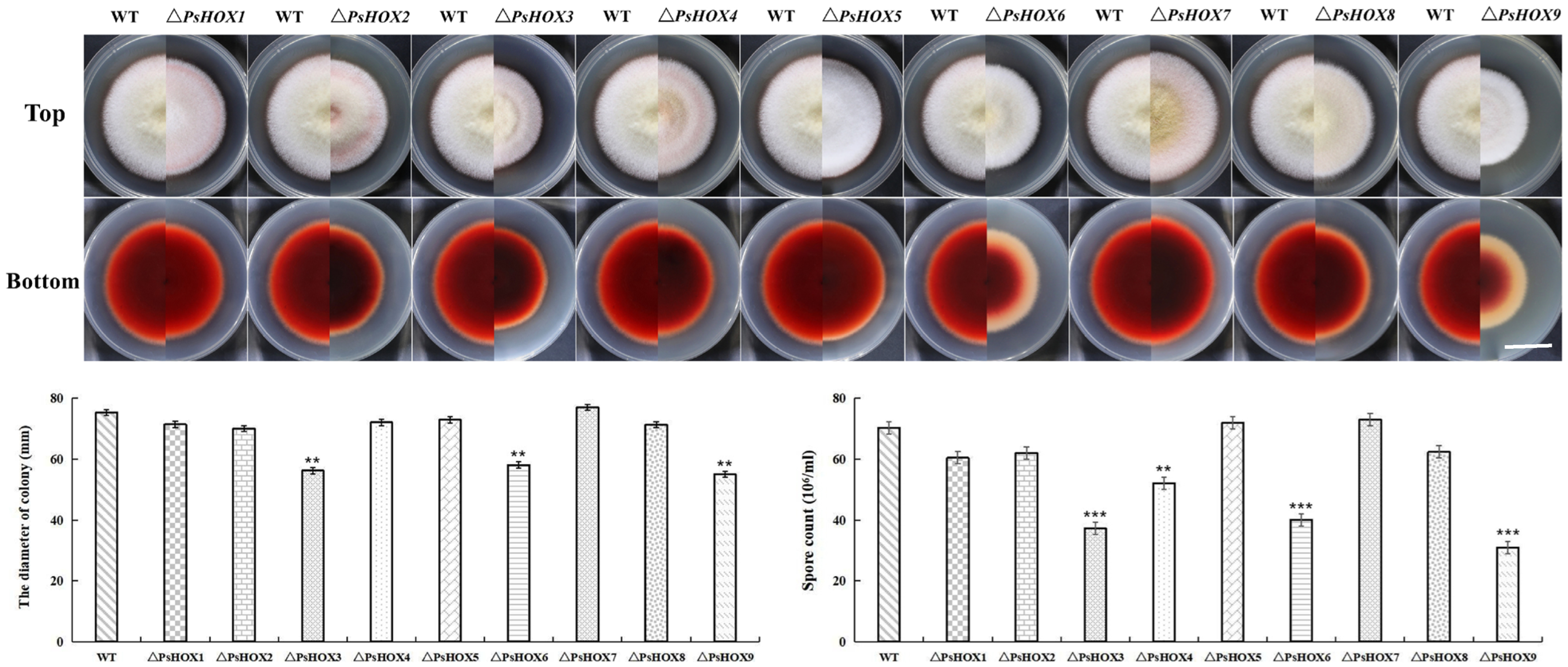
2.6. PsHOX Gene Knockout: Effect on Growth and Conidia Formation
2.7. Knockout of PsHOX Genes for the Pathogenicity of P. sorghina var. saccharum
2.8. PsHOX Gene Knockout: Effect on Secondary Metabolisms of P. sorghina var. saccharum
3. Discussion
4. Materials and Methods
4.1. Isolation of Pathogen Causing Sugarcane Twisted Leaf Disease
4.2. Genome Sequencing, Assembly, and Evaluation
4.3. Gene Prediction and Genome Annotation
4.4. Functional Gene Analysis
4.5. Identification of the PsHOX Family Gene
4.6. Structural Characteristics and Phylogenetic Analysis
4.7. Construction of Homologous Replacement Fragments
4.8. Preparation of Protoplasts
4.9. Transformation
4.10. Screening and Identification of Resistant Transformants
4.11. Complementary Strain Construction
4.12. Phenotypic Observation and Pathogenicity Assay
4.13. LC-MS/MS Method to Detect the Synthesis of Secondary Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bordonal, R.D.O.; Carvalho, J.L.N.; Lal, R.; de Figueiredo, E.B.; De Oliveira, B.G.; Scala, N.L., Jr. Sustainability of sugarcane production in Brazil. A review. Agron. Sustain. Dev. 2018, 38, 13. [Google Scholar] [CrossRef]
- Aveskamp, M.M.; De Gruyter, J.; Crous, P.W. Biology and recent developments in the systematics of Phoma, a complex genus of major quarantine significance. Fungal Divers. 2008, 31, 1–18. [Google Scholar]
- Zaitz, C.; Heins-Vaccari, E.M.; de Freitas, R.S.; Arriagada, G.L.H.; Ruiz, L.; Totoli, S.A.; Marques, A.C.; Rezze, G.G.; Múller, H.; Valente, N.S.; et al. Subcutaneous pheohyphomycosis caused by Phoma cava: Report of a Case and Review of the Literature. Revista do Instituto de Medicina Tropical de São Paulo 1997, 39, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.O.; Gandra, C.R.; Pires, M.F.; Coutinho, S.D.; Castilho, W.; Teixeira, C.M. Survey of bovine mycotic mastitis in dairy herds in the State of São Paulo, Brazil. Mycopathologia 1993, 124, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Y.; Que, Y.X.; Deng, Z.H.; Xu, S.Q.; Rao, G.P.; Zhang, M.Q. First Report of Phoma sp. Causing Twisting and Curling of Crown Leaves of Sugarcane in Mainland of China. Plant Dis. 2014, 98, 850. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wei, J.; Zhang, M.; Xu, S.; Guo, Q.; Wang, X.; Wang, J.; Chen, B.; Que, Y.; Deng, Z.; et al. Identification and Characterization of a New Fungal Pathogen Causing Twisted Leaf Disease of Sugarcane in China. Plant Dis. 2015, 99, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Cellular, genomic and metabolic complexity. Biol. Rev. 2020, 95, 1198–1232. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Yao, W.; Duan, Z.; Powell, C.A.; Chen, B.; Zhang, M. Genome Sequence of Phoma sorghina var. saccharum That Causes Sugarcane Twisted Leaf Disease in China. Mol. Plant-Microbe Interact. 2020, 33, 1092–1094. [Google Scholar] [CrossRef]
- Zhai, Y.; Li, Y.; Zhang, J.; Zhang, Y.; Ren, F.; Zhang, X.; Liu, G.; Liu, X.; Che, Y. Identification of the gene cluster for bistropolone-humulene meroterpenoid biosynthesis in Phoma sp. Fungal Genet. Biol. 2019, 129, 7–15. [Google Scholar] [CrossRef]
- Qin, X.; Luo, Y.; Suh, H.; Wayne, J.; Misulovin, Z.; Roeder, R.; Nussenzweig, M. Transformation by homeobox genes can be mediated by selective transcriptional repression. EMBO J. 1994, 13, 5967–5976. [Google Scholar] [CrossRef] [PubMed]
- Dockx, J.; Quaedvlieg, N.; Keultjes, G.; Kock, P.; Weisbeek, P.; Smeekens, S. The homeobox gene ATK1 of Arabidopsis thaliana is expressed in the shoot apex of the seedling and in flowers and inflorescence stems of mature plants. Plant Mol. Biol. 1995, 28, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Son, S.-H.; Son, Y.-E.; Cho, H.-J.; Chen, W.; Lee, M.-K.; Kim, L.-H.; Han, D.-M.; Park, H.-S. Homeobox proteins are essential for fungal differentiation and secondary metabolism in Aspergillus nidulans. Sci. Rep. 2020, 10, 6094. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, H.; Wang, Y.; Engström, P. DNA-binding and dimerization preferences of Arabidopsis homeodomain-leucine zipper transcription factors in vitro. Plant Mol. Biol. 2001, 45, 63–73. [Google Scholar] [CrossRef]
- Garufi, C.; Brienza, S.; Pugliese, P.; Aschelter, A.M.; A Bensmaine, M.; Bertheault-Cvitkovic, F.; Nisticò, C.; Giunta, S.; Caterino, M.; Giannarelli, D.; et al. Overcoming resistance to chronomodulated 5-fluorouracil and folinic acid by the addition of chronomodulated oxaliplatin in advanced colorectal cancer patients. Anti-Cancer Drugs 2000, 11, 495–501. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, L.; Gong, X.L.; Feng, S.; Liu, X.; Zheng, Y.; Li, Z.; Sun, H.; Wang, D.; Han, J.; et al. Identification of Homeobox Transcription Factor Family in Genome-Wide and Expression Pattern Analysis of the Members in Setosphaeria turcica. Sci. Agric. Sin. 2017, 50, 669–678. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.-Y.; Kim, K.S.; Rho, H.-S.; Chi, M.-H.; Choi, J.; Park, J.; Kong, S.; Park, J.; Goh, J.; et al. Homeobox Transcription Factors Are Required for Conidiation and Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. PLoS Genet. 2009, 5, e1000757. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Izumitsu, K.; Irie, T.; Suzuki, K. The homeobox transcription factor CoHox1 is required for the morphogenesis of infection hyphae in host plants and pathogenicity in Colletotrichum orbiculare. Mycoscience 2018, 60, 110–115. [Google Scholar] [CrossRef]
- Fu, T.; Han, J.-H.; Shin, J.-H.; Song, H.; Ko, J.; Lee, Y.-H.; Kim, K.-T.; Kim, K.S. Homeobox Transcription Factors Are Required for Fungal Development and the Suppression of Host Defense Mechanisms in the Colletotrichum scovillei -Pepper Pathosystem. mBio 2021, 12, e0162021. [Google Scholar] [CrossRef]
- Liu, W.; Xie, S.; Zhao, X.; Chen, X.; Zheng, W.; Lu, G.; Xu, J.-R.; Wang, Z. A Homeobox Gene Is Essential for Conidiogenesis of the Rice Blast Fungus Magnaporthe oryzae. Mol. Plant-Microbe Interact. 2010, 23, 366–375. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, X.; Xie, Q.; Huang, Q.; Zhang, C.; Zhai, H.; Xu, L.; Lu, G.; Shim, W.-B.; Wang, Z. A Conserved Homeobox Transcription Factor Htf1 Is Required for Phialide Development and Conidiogenesis in Fusarium Species. PLoS ONE 2012, 7, e45432. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Wangsanut, T.; Fonzi, W.A.; Rolfes, R.J. TheGRF10homeobox gene regulates filamentous growth in human fungal pathogen Candida albicans. FEMS Yeast Res. 2015, 15, fov093. [Google Scholar] [CrossRef]
- Ferjani, A.; Tsukagoshi, H.; Vassileva, V. Editorial: Model organisms in plant science: Arabidopsis thaliana. Front. Plant Sci. 2023, 14, 1279230. [Google Scholar] [CrossRef]
- McConnell, J.R.; Emery, J.; Eshed, Y.; Bao, N.; Bowman, J.; Barton, M.K. Role of PHABULOSA and PHAVOLUTA in determining radial patterning in shoots. Nature 2001, 411, 709–713. [Google Scholar] [CrossRef]
- Ochando, I.; Jover-Gil, S.; Ripoll, J.J.; Candela, H.; Vera, A.; Ponce, M.R.; Martínez-Laborda, A.; Micol, J.L. Mutations in the MicroRNA Complementarity Site of the INCURVATA4 Gene Perturb Meristem Function and Adaxialize Lateral Organs in Arabidopsis. Plant Physiol. 2006, 141, 607–619. [Google Scholar] [CrossRef]
- Wang, S.; Pan, K.Y.; Yao, Z.T.; Yao, W.; Zhang, M.Q. Construction of gene knockout system in sugarcane pokkah boeng pathogen Fusarium proloferatum YN41 based on transformation of protoplast. Acta Phytopathol. Sin. 2023, 53, 72–83. [Google Scholar] [CrossRef]
- Gubenko, I.S. Drosophila genes that encode LIM-domain containing proteins: Their organization, functions, and interactions. Tsitologiia I Genet. 2006, 40, 44–67. [Google Scholar]
- Jasinski, S.; Kaur, H.; Tattersall, A.; Tsiantis, M. Negative regulation of KNOX expression in tomato leaves. Planta 2007, 226, 1255–1263. [Google Scholar] [CrossRef]
- Casselton, L.A.; Olesnicky, N.S. Molecular Genetics of Mating Recognition in Basidiomycete Fungi. Microbiol. Mol. Biol. Rev. 1998, 62, 55–70. [Google Scholar] [CrossRef]
- Bürglin, T.R.; Affolter, M. Homeodomain proteins: An update. Chromosoma 2015, 125, 497–521. [Google Scholar] [CrossRef]
- Gehring, W.J.; Hiromi, Y. Homeotic Genes and the Homeobox. Annu. Rev. Genet. 1986, 20, 147–173. [Google Scholar] [CrossRef]
- Zheng, W.H.; Zhao, X.; Zhang, C.K.; Lu, G.D.; Wang, Z.H.; Wang, A.R. Gene structure and phylogenetic relationships of Homeobox transcriptional factors in Fusarium graminearum. Chin. J. Trop. Crops. 2009, 30, 205–210. [Google Scholar]
- Xiong, D.; Wang, Y.; Deng, C.; Hu, R.; Tian, C. Phylogenic analysis revealed an expanded C2H2-homeobox subfamily and expression profiles of C2H2 zinc finger gene family in Verticillium dahliae. Gene 2015, 562, 169–179. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, Y.-H. Gene Expression Profiling during Conidiation in the Rice Blast Pathogen Magnaporthe oryzae. PLoS ONE 2012, 7, e43202. [Google Scholar] [CrossRef]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The Stem Cell Population of Arabidopsis Shoot Meristems Is Maintained by a Regulatory Loop between the CLAVATA and WUSCHEL Genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef]
- Arnaise, S.; Zickler, D.; Poisier, C.; Debuchy, R. pah1: A homeobox gene involved in hyphal morphology and microconidiogenesis in the filamentous ascomycete Podospora anserina. Mol. Microbiol. 2001, 39, 54–64. [Google Scholar] [CrossRef]
- Argüelles, J.C. Physiological roles of trehalose in bacteria and yeasts: A comparative analysis. Arch. Microbiol. 2000, 174, 217–224. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Liu, X.; Wen, H.; Wang, M. A simple method of genomic DNA extraction suitable for analysis of bulk fungal strains. Lett. Appl. Microbiol. 2010, 51, 114–118. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef]
- Seberg, O.; Petersen, G. A unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat. Rev. Genet. 2009, 10, 276. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4–10. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, 215–225. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 995–1001. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Green, P.J.; Richardson, S. Hidden Markov Models and Disease Mapping. J. Am. Stat. Assoc. 2002, 97, 1055–1070. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Saier, M.H. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef]
- Winnenburg, R. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Locus | Number of Amino Acids | Molecular Weight | Theoretical PI | Instability Index | Stability | Sub-Cellular Localization |
|---|---|---|---|---|---|---|---|---|
| PsHOX1 | PHOM_00410 | Contig04: 763,773–766,172 | 783 | 85,593.90 | 6.39 | 61.36 | unstable | Nucleus |
| PsHOX2 | PHOM_01626 | Contig05: 386,206–388,014 | 532 | 57,846.51 | 8.65 | 67.96 | unstable | Nucleus |
| PsHOX3 | PHOM_02470 | Contig19: 726,424–728,280 | 583 | 63,996.62 | 6.38 | 59.55 | unstable | Nucleus |
| PsHOX4 | PHOM_03333 | Contig00: 1,136,679–1,138,132 | 448 | 50,586.42 | 8.84 | 72.50 | unstable | Nucleus or Mitochondrial |
| PsHOX5 | PHOM_04409 | Contig02: 1,641,196–1,642,806 | 521 | 59,044.87 | 6.66 | 52.41 | unstable | Nucleus |
| PsHOX6 | PHOM_05072 | Contig02: 2,122,838–2,125,523 | 878 | 99,276.85 | 6.63 | 57.18 | unstable | Nucleus |
| PsHOX7 | PHOM_05197 | Contig03: 190,707–194,407 | 1160 | 133,759.79 | 5.35 | 60.17 | unstable | Nucleus |
| PsHOX8 | PHOM_06420 | Contig02: 1,775,319–1,778,410 | 882 | 98,336.46 | 5.85 | 53.37 | unstable | Nucleus |
| PsHOX9 | PHOM_07085 | Contig00: 2,533,344–2,537,402 | 963 | 108,861.84 | 7.20 | 55.94 | unstable | Nucleus |
| Categories | Number | Secondary Metabolites |
|---|---|---|
| Benzene and substituted derivatives | 10 | 4-Dodecylbenzenesulfonic acid; 3-Phenoxybenzoic acid; 3-Aminosalicylic acid; Gentian violet; 2,4-Dimethylbenzaldehyde; Benzylamine; Monobutyl phthalate; N,N’-Diphenylurea; Terephthalic acid; Dibutyl phthalate |
| Carboxylic acids and derivatives | 15 | l-Glutamic acid; l-Norleucine; l-Phenylalanine; l-Pyroglutamic acid; l-Ergothioneine; l-Histidine; N6-Acetyl-l-lysine; l-Cysteine; Citric acid; Valylproline; 4-Guanidinobutyric acid; l-Tyrosine; 4-Acetamidobutanoic acid; N-Acetyl-l-leucine; Isoleucine |
| Fatty Acyls | 16 | 16-Hydroxyhexadecanoic acid; Oleic acid; Stearic Acid; Palmitic Acid; Linoleic Acid; Palmitoleic acid; Azelaic acid; Nonanoic acid; Myristic Acid; Arachidonic acid; Docosahexaenoic Acid; Dimethyl succinate; Oleamide; Acetyl-l-carnitine; Hexadecanamide; Propionylcarnitine |
| Organooxygen compounds | 13 | α,α-Trehalose; Choline; α-d-Mannose 1-phosphate; Dulcitol; d-Raffinose; d-Ribose-1-phosphate; Glucose 1-phosphate; Triethanolamine; Spermidine; Cyclohexylamine; Procyclidine; Muscone; 2-Acetylpyridine |
| Pyridines and derivatives | 7 | Picolinic acid; Nicotinamide; Nicotinic acid; 6-Hydroxynicotinic acid; Pyridoxamine; Pyridoxine; 6-Methylquinoline |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, Y.; Deng, J.; Akbar, S.; Duan, Z.; Zhang, C.; Lin, W.; Wu, S.; Yue, Y.; Yao, W.; Xu, J.; et al. Genome-Wide Identification and Characterization of Homeobox Transcription Factors in Phoma sorghina var. saccharum Causing Sugarcane Twisted Leaf Disease. Int. J. Mol. Sci. 2024, 25, 5346. https://doi.org/10.3390/ijms25105346
Bao Y, Deng J, Akbar S, Duan Z, Zhang C, Lin W, Wu S, Yue Y, Yao W, Xu J, et al. Genome-Wide Identification and Characterization of Homeobox Transcription Factors in Phoma sorghina var. saccharum Causing Sugarcane Twisted Leaf Disease. International Journal of Molecular Sciences. 2024; 25(10):5346. https://doi.org/10.3390/ijms25105346
Chicago/Turabian StyleBao, Yixue, Jinlan Deng, Sehrish Akbar, Zhenzhen Duan, Chi Zhang, Wenfeng Lin, Suyan Wu, Yabing Yue, Wei Yao, Jianlong Xu, and et al. 2024. "Genome-Wide Identification and Characterization of Homeobox Transcription Factors in Phoma sorghina var. saccharum Causing Sugarcane Twisted Leaf Disease" International Journal of Molecular Sciences 25, no. 10: 5346. https://doi.org/10.3390/ijms25105346
APA StyleBao, Y., Deng, J., Akbar, S., Duan, Z., Zhang, C., Lin, W., Wu, S., Yue, Y., Yao, W., Xu, J., & Zhang, M. (2024). Genome-Wide Identification and Characterization of Homeobox Transcription Factors in Phoma sorghina var. saccharum Causing Sugarcane Twisted Leaf Disease. International Journal of Molecular Sciences, 25(10), 5346. https://doi.org/10.3390/ijms25105346