


Cytoskeleton Rearrangement in Podocytopathies: An Update

Abstract
1. Introduction
2. Overview of Podocytopathies
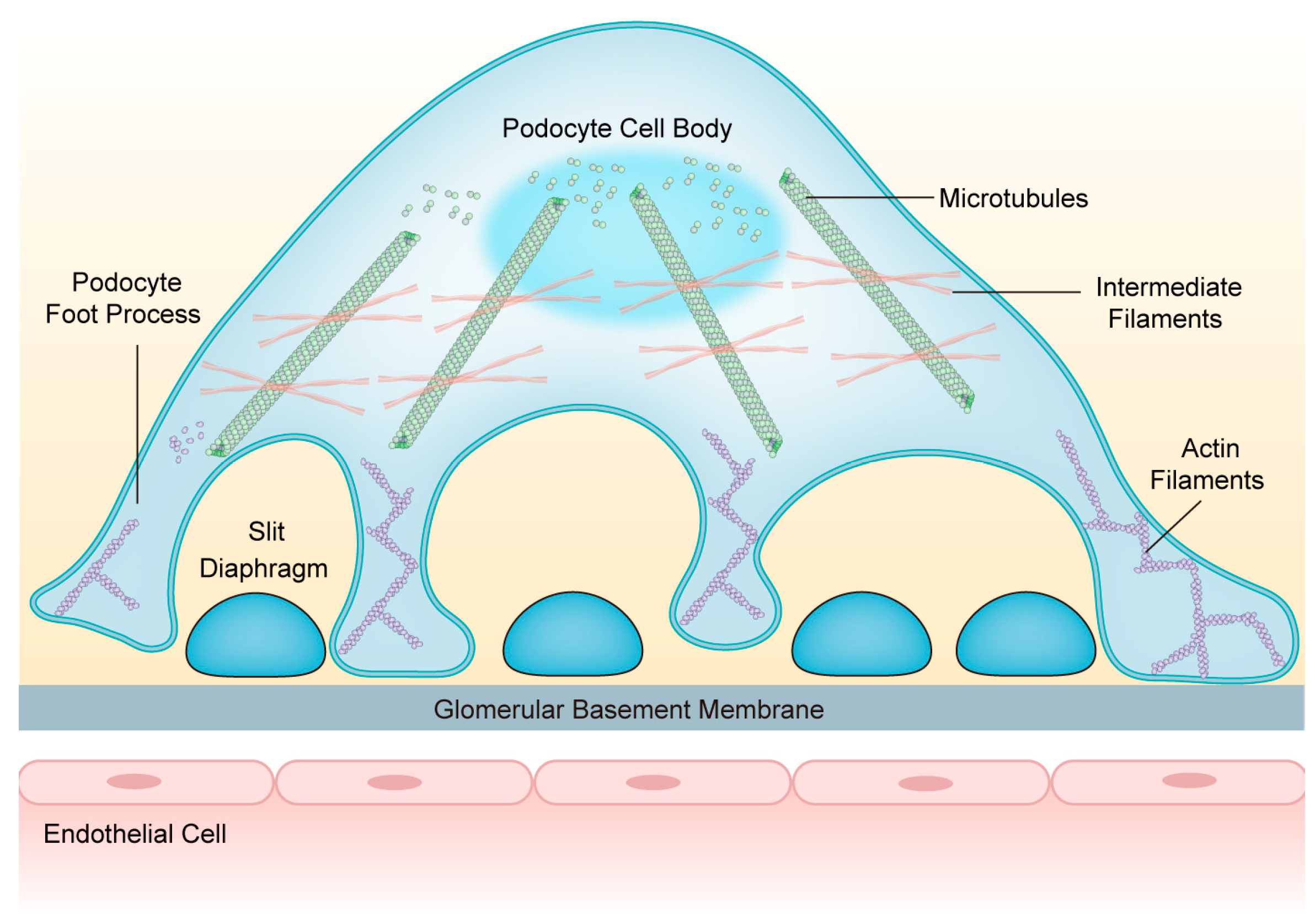
3. Composition and Structure of the Podocyte Cytoskeleton
3.1. Actin Cytoskeleton
3.2. Microtubules
3.3. Intermediate Filaments
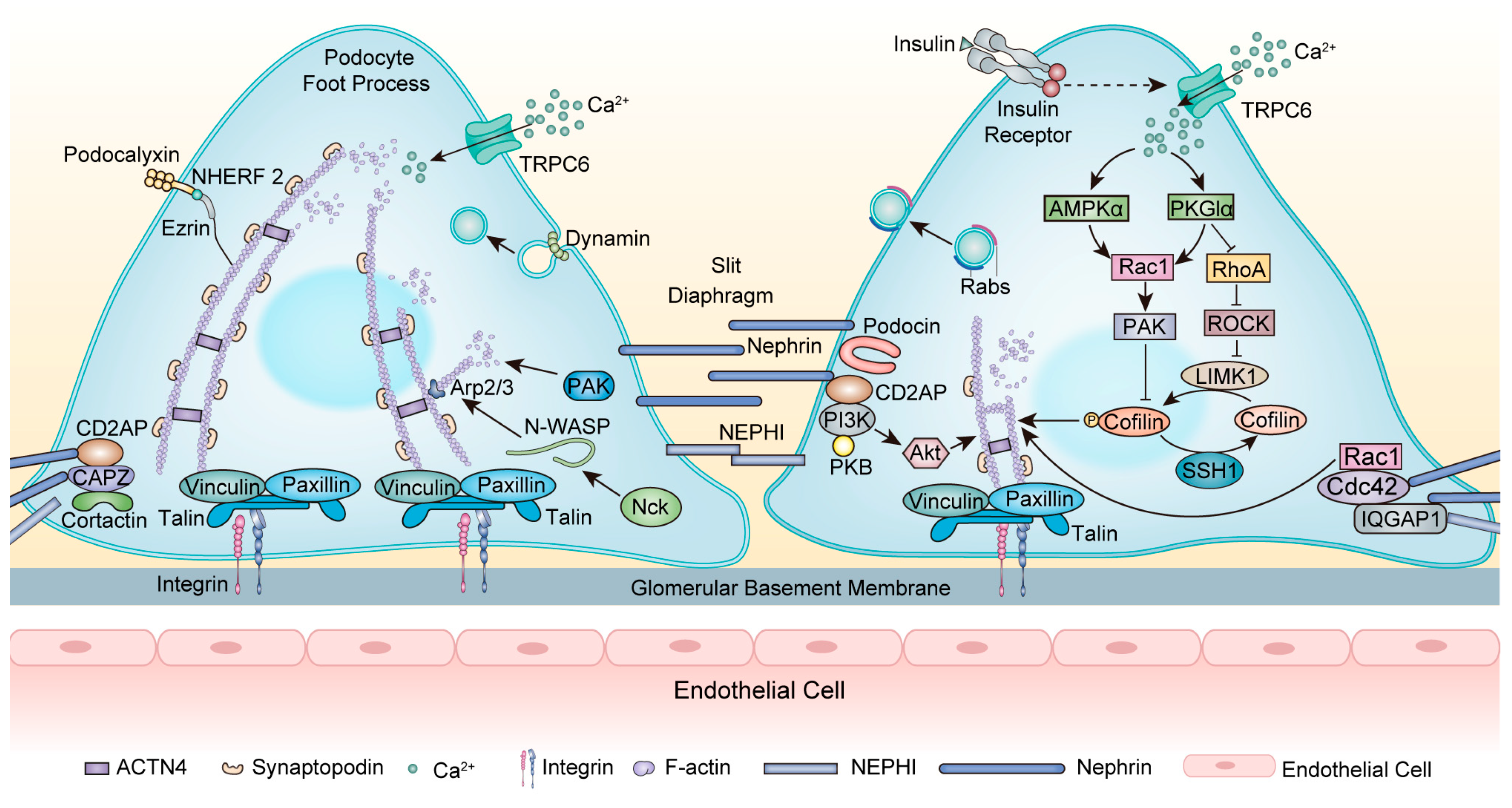
4. Cytoskeleton Rearrangement in Podocytopathies and Podocyte-Centric Diseases
4.1. Focal Segmental Glomerulosclerosis
4.2. Minimal Change Disease
4.3. Diabetic Kidney Disease
4.4. Membranous Nephropathy
4.5. Lupus Nephritis
5. Therapeutic Implications
5.1. Glucocorticoids
5.2. Rituximab
5.3. Calcineurin Inhibitor
5.4. Abatacept
5.5. Others
5.6. New Potential Therapeutic Strategies

{kind=link}
{kind=link}
| Agents | Medicine | Mechanisms/Pathways | References |
|---|---|---|---|
| GCs | DEX | Upregulating nephrin and increasing nephrin phosphorylation | [132,136,137] |
| Facilitating nephrin synthesis and transporting nephrin to the plasma membrane | [133] | ||
| Blocking TRPC6 signaling and stabilizing TRPC6 expression | [138] | ||
| Stabilizing ACTN4 expression | [139] | ||
| Inhibiting Rac1 overactivity | [140] | ||
| Increasing synaptopodin expression | [142] | ||
| / | RTX | Preventing reduced expression of nephrin and podocin | [149] |
| Stabilizing SMPDL-3b | [152] | ||
| CNIs | CsA | Maintaining synaptopodin by blocking the calcineurin-mediated dephosphorylation of synaptopodin | [161] |
| Downregulating TRPC6 expression | [166] | ||
| Decreasing WAVE1 expression | [168] | ||
| TAC | Reducing Angptl4 expression | [120,170] | |
| Increasing FKBP12 expression and enhancing the interaction of FKBP12 with 14-3-3β and synaptopodin | [171] | ||
| Increasing nephrin and podocin expression and maintaining MAP1 LC3 | [172,173] | ||
| / | Abatacept | Stabilizing β1 integrin activation and its association with talin | [178] |
| / | Bis-T-23 | Promoting actin-dependent dynamin oligomerization | [195,196,197] |
| Rho kinase inhibitor | Fasudi | Modulating nephrin and CD2AP | [200] |
| Inhibiting CaMK4/RhoA signaling and inducing the release of YAP | [201] | ||
| Y27632 | Stabilizing actin cytoskeleton and MTs | [202,203] | |
| RAS inhibitor | ACEI/ARB | Stabilizing nephrin expression | [204,205] |
| Inhibiting TRPC6 expression | [207] | ||
| mTOR-inhibitor | EV | Preventing inhibition of the RhoA–ROCK–MLC pathway Stabilizing MTs | [208,209] |
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Fogo, A.B. The Targeted Podocyte. J. Clin. Investig. 2011, 121, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Imasawa, T.; Rossignol, R. Podocyte Energy Metabolism and Glomerular Diseases. Int. J. Biochem. Cell Biol. 2013, 45, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Durvasula, R.V.; Shankland, S.J. Podocyte Injury and Targeting Therapy: An Update. Curr. Opin. Nephrol. Hypertens. 2006, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J.; Dylewski, J. Regulation of the Actin Cytoskeleton in Podocytes. Cells 2020, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Altintas, M.M. Podocytes. F1000Research 2016, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Lan, B.; Liu, H.; Persson, P.B.; Lai, E.Y.; Mao, J. A Critical Role of the Podocyte Cytoskeleton in the Pathogenesis of Glomerular Proteinuria and Autoimmune Podocytopathies. Acta Physiol. 2022, 235, e13850. [Google Scholar] [CrossRef]
- Endlich, N.; Siegerist, F.; Endlich, K. Are Podocytes Motile? Pflug. Arch. Eur. J. Physiol. 2017, 469, 951–957. [Google Scholar] [CrossRef]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte—Actin Dynamics in Health and Disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef]
- Lappalainen, P.; Kotila, T.; Jégou, A.; Romet-Lemonne, G. Biochemical and Mechanical Regulation of Actin Dynamics. Nat. Rev. Mol. Cell Biol. 2022, 23, 836–852. [Google Scholar] [CrossRef]
- Welsh, G.I.; Saleem, M.A. The Podocyte Cytoskeleton—Key to a Functioning Glomerulus in Health and Disease. Nat. Rev. Nephrol. 2012, 8, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Ahn, W.; Bomback, A.S. Approach to Diagnosis and Management of Primary Glomerular Diseases Due to Podocytopathies in Adults: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.; Lugli, G.; Raglianti, V.; Ravaglia, F.; Buti, E.; Landini, S.; Becherucci, F. Defining Diagnostic Trajectories in Patients with Podocytopathies. Clin. Kidney J. 2022, 15, 2006–2019. [Google Scholar] [CrossRef] [PubMed]
- Barisoni, L.; Schnaper, H.W.; Kopp, J.B. A Proposed Taxonomy for the Podocytopathies: A Reassessment of the Primary Nephrotic Diseases. Clin. J. Am. Soc. Nephrol. 2007, 2, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ishibe, S. Targeting the Podocyte Cytoskeleton: From Pathogenesis to Therapy in Proteinuric Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 1577–1583. [Google Scholar] [CrossRef]
- Pollard, T.D.; Goldman, R.D. Overview of the Cytoskeleton from an Evolutionary Perspective. Cold Spring Harb. Perspect. Biol. 2018, 10, a030288. [Google Scholar] [CrossRef]
- Muraleedharan, S.; Sam, A.; Skaer, H.; Inamdar, M.S. Networks That Link Cytoskeletal Regulators and Diaphragm Proteins Underpin Filtration Function in Drosophila Nephrocytes. Exp. Cell Res. 2018, 364, 234–242. [Google Scholar] [CrossRef]
- Ahmadian, E.; Eftekhari, A.; Atakishizada, S.; Valiyeva, M.; Ardalan, M.; Khalilov, R.; Kavetskyy, T. Podocytopathy: The Role of Actin Cytoskeleton. Biomed. Pharmacother. 2022, 156, 113920. [Google Scholar] [CrossRef]
- Qu, C.; Roth, R.; Puapatanakul, P.; Loitman, C.; Hammad, D.; Genin, G.M.; Miner, J.H.; Suleiman, H.Y. Three-Dimensional Visualization of the Podocyte Actin Network Using Integrated Membrane Extraction, Electron Microscopy, and Machine Learning. J. Am. Soc. Nephrol. 2022, 33, 155–173. [Google Scholar] [CrossRef]
- Xu, W.; Ge, Y.; Liu, Z.; Gong, R. Glycogen Synthase Kinase 3β Orchestrates Microtubule Remodeling in Compensatory Glomerular Adaptation to Podocyte Depletion. J. Biol. Chem. 2015, 290, 1348–1363. [Google Scholar] [CrossRef] [PubMed]
- Gödel, M.; Temerinac, D.; Grahammer, F.; Hartleben, B.; Kretz, O.; Riederer, B.M.; Propst, F.; Kohl, S.; Huber, T.B. Microtubule Associated Protein 1b (MAP1B) Is a Marker of the Microtubular Cytoskeleton in Podocytes but Is Not Essential for the Function of the Kidney Filtration Barrier in Mice. PLoS ONE 2015, 10, e0140116. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Mundel, P. A Role of Microtubules during the Formation of Cell Processes in Neuronal and Non-Neuronal Cells. Cell Tissue Res. 1998, 291, 163–174. [Google Scholar] [CrossRef]
- Kobayashi, N.; Gao, S.; Chen, J.; Saito, K.; Miyawaki, K.; Li, C.; Pan, L.; Saito, S.; Terashita, T.; Matsuda, S. Process Formation of the Renal Glomerular Podocyte: Is There Common Molecular Machinery for Processes of Podocytes and Neurons? Anat. Sci. Int. 2004, 79, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Heid, H.W.; Sakai, T.; Kriz, W.; Huber, G.; Mundel, P. Molecular Characterization Reveals Identity of Microtubule-Associated Proteins MAP3 and MAP4. Biochem. Biophys. Res. Commun. 2000, 268, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, A.; Bucci, C. Role of Intermediate Filaments in Vesicular Traffic. Cells 2016, 5, 20. [Google Scholar] [CrossRef]
- Zou, J.; Yaoita, E.; Watanabe, Y.; Yoshida, Y.; Nameta, M.; Li, H.; Qu, Z.; Yamamoto, T. Upregulation of Nestin, Vimentin, and Desmin in Rat Podocytes in Response to Injury. Virchows Arch. 2006, 448, 485–492. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, T.; Yu, X.; Muwonge, A.N.; Anandakrishnan, N.; Wong, N.J.; Haydak, J.C.; Reid, J.M.; Fu, J.; Wong, J.S.; et al. LIM-Nebulette Reinforces Podocyte Structural Integrity by Linking Actin and Vimentin Filaments. J. Am. Soc. Nephrol. 2020, 31, 2372–2391. [Google Scholar] [CrossRef]
- Embry, A.E.; Mohammadi, H.; Niu, X.; Liu, L.; Moe, B.; Miller-Little, W.A.; Lu, C.Y.; Bruggeman, L.A.; McCulloch, C.A.; Janmey, P.A.; et al. Biochemical and Cellular Determinants of Renal Glomerular Elasticity. PLoS ONE 2016, 11, e0167924. [Google Scholar] [CrossRef]
- Chen, J.; Boyle, S.; Zhao, M.; Su, W.; Takahashi, K.; Davis, L.; DeCaestecker, M.; Takahashi, T.; Breyer, M.D.; Hao, C.-M. Differential Expression of the Intermediate Filament Protein Nestin during Renal Development and Its Localization in Adult Podocytes. J. Am. Soc. Nephrol. 2006, 17, 1283–1291. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Veissi, S.T.; Smeets, B.; van Wijk, J.A.E.; Classens, R.; van der Velden, T.J.A.M.; Jeronimus-Klaasen, A.; Veltkamp, F.; Nienhuis, E.M.; Morello, W.; Montini, G.; et al. Circulating Permeability Factors in Focal Segmental Glomerulosclerosis: In Vitro Detection. Kidney Int. Rep. 2022, 7, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Massengill, S.; Trachtman, H. Genetic Spectrum of Nephrotic Syndrome: Impact of Podocytopathy in Adult Life. Adv. Chronic Kidney Dis. 2022, 29, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Patrakka, J.; Tryggvason, K. Nephrin—A Unique Structural and Signaling Protein of the Kidney Filter. Trends Mol. Med. 2007, 13, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Butt, L.; Unnersjö-Jess, D.; Höhne, M.; Edwards, A.; Binz-Lotter, J.; Reilly, D.; Hahnfeldt, R.; Ziegler, V.; Fremter, K.; Rinschen, M.M.; et al. A Molecular Mechanism Explaining Albuminuria in Kidney Disease. Nat. Metab. 2020, 2, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Santín, S.; García-Maset, R.; Ruíz, P.; Giménez, I.; Zamora, I.; Peña, A.; Madrid, Á.; Camacho, J.A.; Fraga, G.; Sánchez-Moreno, A.; et al. Nephrin Mutations Cause Childhood- and Adult-Onset Focal Segmental Glomerulosclerosis. Kidney Int. 2009, 76, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, Y.; Liu, L.; Hosoyamada, M.; Endou, H.; Kudo, A.; Takenaka, H.; Higashihara, E.; Bessho, F.; Takahashi, S.; Kershaw, D.; et al. Disease-Causing Missense Mutations in NPHS2 Gene Alter Normal Nephrin Trafficking to the Plasma Membrane. Kidney Int. 2004, 66, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Riguetti, M.T.P.; Varela, P.; Fernandes, D.E.; Polito, M.G.; Casimiro, F.M.; Pesquero, J.B.; Mastroianni-Kirsztajn, G. Familial Focal Segmental Glomerulosclerosis With Late-Onset Presentation and R229Q/R291W Podocin Mutations. Front. Genet. 2020, 11, 533373. [Google Scholar] [CrossRef]
- Park, H.-Y.; Seong, S.-B.; Min, S.-Y.; Ha, T.-S. CD2-Associated Protein/Phosphoinositide 3-Kinase Signaling Has a Preventive Role in Angiotensin II-Induced Podocyte Apoptosis. Int. J. Biochem. Cell Biol. 2016, 79, 370–381. [Google Scholar] [CrossRef]
- Takano, T.; Bareke, E.; Takeda, N.; Aoudjit, L.; Baldwin, C.; Pisano, P.; Matsuda, J.; El Andalousi, J.; Muhtadie, L.; Bernard, C.; et al. Recessive Mutation in CD2AP Causes Focal Segmental Glomerulosclerosis in Humans and Mice. Kidney Int. 2019, 95, 57–61. [Google Scholar] [CrossRef]
- Michaud, J.-L.R.; Hosseini-Abardeh, M.; Farah, K.; Kennedy, C.R.J. Modulating α-Actinin-4 Dynamics in Podocytes. Cell Motil. Cytoskelet. 2009, 66, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Odenthal, J.; Dittrich, S.; Ludwig, V.; Merz, T.; Reitmeier, K.; Reusch, B.; Höhne, M.; Cosgun, Z.C.; Hohenadel, M.; Putnik, J.; et al. Modeling of ACTN4-Based Podocytopathy Using Drosophila Nephrocytes. Kidney Int. Rep. 2023, 8, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Blasutig, I.M.; Eremina, V.; Ruston, J.M.; Bladt, F.; Li, H.; Huang, H.; Larose, L.; Li, S.S.-C.; Takano, T.; et al. Nck Adaptor Proteins Link Nephrin to the Actin Cytoskeleton of Kidney Podocytes. Nature 2006, 440, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Phippen, N.J.; Keyvani Chahi, A.; Tilak, M.; Banerjee, S.L.; Lu, P.; New, L.A.; Williamson, C.R.; Platt, M.J.; Simpson, J.A.; et al. Complementary Nck1/2 Signaling in Podocytes Controls α Actinin-4–Mediated Actin Organization, Adhesion, and Basement Membrane Composition. J. Am. Soc. Nephrol. 2022, 33, 1546–1567. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.; Reidy, K.J. Genetic Causes of Proteinuria and Nephrotic Syndrome: Impact on Podocyte Pathobiology. Pediatr. Nephrol. 2015, 30, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Brähler, S.; Yu, H.; Suleiman, H.; Krishnan, G.M.; Saunders, B.T.; Kopp, J.B.; Miner, J.H.; Zinselmeyer, B.H.; Shaw, A.S. Intravital and Kidney Slice Imaging of Podocyte Membrane Dynamics. J. Am. Soc. Nephrol. 2016, 27, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin Protects Against Proteinuria by Disrupting Cdc42:IRSp53:Mena Signaling Complexes in Kidney Podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Suleiman, H.Y.; Miner, J.H. Synaptopodin Is Dispensable for Normal Podocyte Homeostasis but Is Protective in the Context of Acute Podocyte Injury. J. Am. Soc. Nephrol. 2020, 31, 2815–2832. [Google Scholar] [CrossRef]
- Li, Q.; Gulati, A.; Lemaire, M.; Nottoli, T.; Bale, A.; Tufro, A. Rho-GTPase Activating Protein Myosin MYO9A Identified as a Novel Candidate Gene for Monogenic Focal Segmental Glomerulosclerosis. Kidney Int. 2021, 99, 1102–1117. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, L.; Chen, Y.; Zhang, H.; Yu, C.; Zhou, F.; Zhang, Z.; Jiang, L.; Li, R.; Ma, J.; et al. RhoA Deficiency Disrupts Podocyte Cytoskeleton and Induces Podocyte Apoptosis by Inhibiting YAP/Dendrin Signal. BMC Nephrol. 2016, 17, 66. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, L.; Chen, Y.; Zhang, H.; Zhang, Q.; Li, R.; Ma, J.; Li, Z.; Yu, C.; Lai, Y.; et al. Cdc42 Deficiency Induces Podocyte Apoptosis by Inhibiting the Nwasp/Stress Fibers/YAP Pathway. Cell Death Dis. 2016, 7, e2142. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arroyo, O.; Selma-Soriano, E.; Ortega, A.; Cortes, R.; Redon, J. Small Rab GTPases in Intracellular Vesicle Trafficking: The Case of Rab3A/Raphillin-3A Complex in the Kidney. Int. J. Mol. Sci. 2021, 22, 7679. [Google Scholar] [CrossRef] [PubMed]
- Rastaldi, M.P.; Armelloni, S.; Berra, S.; Li, M.; Pesaresi, M.; Poczewski, H.; Langer, B.; Kerjaschki, D.; Henger, A.; Blattner, S.M.; et al. Glomerular Podocytes Possess the Synaptic Vesicle Molecule Rab3A and Its Specific Effector Rabphilin-3a. Am. J. Pathol. 2003, 163, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Selma-Soriano, E.; Llamusi, B.; Fernández-Costa, J.M.; Ozimski, L.L.; Artero, R.; Redón, J. Rabphilin Involvement in Filtration and Molecular Uptake in Drosophila Nephrocytes Suggests a Similar Role in Human Podocytes. Dis. Models Mech. 2020, 13, dmm041509. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Staruschenko, A. TRPC6 Channel as an Emerging Determinant of the Podocyte Injury Susceptibility in Kidney Diseases. Am. J. Physiol.-Ren. Physiol. 2015, 309, F393–F397. [Google Scholar] [CrossRef]
- Jiang, L.; Ding, J.; Tsai, H.; Li, L.; Feng, Q.; Miao, J.; Fan, Q. Over-Expressing Transient Receptor Potential Cation Channel 6 in Podocytes Induces Cytoskeleton Rearrangement through Increases of Intracellular Ca2+ and RhoA Activation. Exp. Biol. Med. 2011, 236, 184–193. [Google Scholar] [CrossRef]
- Shalygin, A.; Shuyskiy, L.S.; Bohovyk, R.; Palygin, O.; Staruschenko, A.; Kaznacheyeva, E. Cytoskeleton Rearrangements Modulate TRPC6 Channel Activity in Podocytes. Int. J. Mol. Sci. 2021, 22, 4396. [Google Scholar] [CrossRef]
- Riehle, M.; Büscher, A.K.; Gohlke, B.-O.; Kaßmann, M.; Kolatsi-Joannou, M.; Bräsen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.-Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef]
- Beaudreuil, S.; Zhang, X.; Herr, F.; Harper, F.; Candelier, J.J.; Fan, Y.; Yeter, H.; Dudreuilh, C.; Lecru, L.; Vazquez, A.; et al. Circulating CASK Is Associated with Recurrent Focal Segmental Glomerulosclerosis after Transplantation. PLoS ONE 2019, 14, e0219353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y.; Zhao, X. FAM40A Alters the Cytoskeleton of Podocytes in Familial Focal and Segmental Glomerulosclerosis by Regulating F-Actin and Nephrin. Arch. Med. Sci. 2019, 15, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’Agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of Decay-Accelerating Factor Triggers Podocyte Injury and Glomerulosclerosis. J. Exp. Med. 2020, 217, e20191699. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Wang, X.; Yin, X.; Li, Y.; Liu, X.; Wang, H.; Liu, X.; Zhang, J.; Gao, H.; Shi, B.; et al. Plectin Protects Podocytes from Adriamycin-induced Apoptosis and F-actin Cytoskeletal Disruption through the Integrin A6β4/FAK/P38 MAPK Pathway. J. Cell. Mol. Medi 2018, 22, 5450–5467. [Google Scholar] [CrossRef] [PubMed]
- Solberg, R.; Lunde, N.N.; Forbord, K.M.; Okla, M.; Kassem, M.; Jafari, A. The Mammalian Cysteine Protease Legumain in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15983. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Matthews, S.P.; Reinheckel, T.; Fleming, S.; Watts, C. Asparagine Endopeptidase Is Required for Normal Kidney Physiology and Homeostasis. FASEB J. 2011, 25, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Yamasaki, M.; Fukui, T. Degradation of Acetoacetyl-CoA Synthetase, a Ketone Body-Utilizing Enzyme, by Legumain in the Mouse Kidney. Biochem. Biophys. Res. Commun. 2014, 453, 631–635. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J.; Prescott, A.; Van Kasteren, S.; Pedrioli, D.L.; McLean, I.; Moles, A.; Reinheckel, T.; Poli, V.; Watts, C. Lysosomal Protease Deficiency or Substrate Overload Induces an Oxidative-Stress Mediated STAT3-Dependent Pathway of Lysosomal Homeostasis. Nat. Commun. 2018, 9, 5343. [Google Scholar] [CrossRef]
- Yamamoto-Nonaka, K.; Koike, M.; Asanuma, K.; Takagi, M.; Oliva Trejo, J.A.; Seki, T.; Hidaka, T.; Ichimura, K.; Sakai, T.; Tada, N.; et al. Cathepsin D in Podocytes Is Important in the Pathogenesis of Proteinuria and CKD. J. Am. Soc. Nephrol. 2016, 27, 2685–2700. [Google Scholar] [CrossRef]
- Qiu, Y.; Lei, C.; Zeng, J.; Xie, Y.; Cao, Y.; Yuan, Q.; Su, H.; Zhang, Z.; Zhang, C. Asparagine Endopeptidase Protects Podocytes in Adriamycin-Induced Nephropathy by Regulating Actin Dynamics through Cleaving Transgelin. Mol. Ther. 2023, 31, 3337–3354. [Google Scholar] [CrossRef]
- Vivarelli, M.; Massella, L.; Ruggiero, B.; Emma, F. Minimal Change Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Uchida, K.; Uchida, S.; Sasaki, S.; Nitta, K. Dexamethasone Increases the Phosphorylation of Nephrin in Cultured Podocytes. Clin. Exp. Nephrol. 2011, 15, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Kilpeläinen, P.; Hellman, U.; Sun, Y.; Wartiovaara, J.; Morgunova, E.; Pikkarainen, T.; Yan, K.; Jonsson, A.P.; Tryggvason, K. Characterization of the Interactions of the Nephrin Intracellular Domain: Evidence that the Scaffolding Protein IQGAP1 Associates with Nephrin. FEBS J. 2004, 272, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, W.; Yang, Y.; Pan, Y.; Yang, Q.; Chen, X.; Singhal, P.C.; Ding, G. IQGAP1 Regulates Actin Cytoskeleton Organization in Podocytes through Interaction with Nephrin. Cell. Signal. 2015, 27, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, Q.; Zhong, Z.; Liang, W.; Zhang, L.; Yang, Y.; Ding, G. Role of C-Abl and Nephrin in Podocyte Cytoskeletal Remodeling Induced by Angiotensin II. Cell Death Dis. 2018, 9, 185. [Google Scholar] [CrossRef]
- Ha, T.-S.; Nam, J.A.; Seong, S.-B.; Saleem, M.A.; Park, S.J.; Shin, J.I. Montelukast Improves the Changes of Cytoskeletal and Adaptor Proteins of Human Podocytes by Interleukin-13. Inflamm. Res. 2017, 66, 793–802. [Google Scholar] [CrossRef]
- Peng, L.; Zhao, H.; Liu, S.; Yuan, Y.; Yuan, C.; Mwamunyi, M.; Pearce, D.; Yao, L. Lack of Serum- and Glucocorticoid-inducible Kinase 3 Leads to Podocyte Dysfunction. FASEB J. 2018, 32, 576–587. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Z.; Deng, H.; Liu, M.; Lin, X.; Zhang, M.; Li, G.; Yue, S.; Gao, X. ANGPTL4 Promotes Nephrotic Syndrome by Downregulating Podocyte Expression of ACTN4 and Podocin. Biochem. Biophys. Res. Commun. 2023, 639, 176–182. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, Y.; Lin, C.; Weng, C.; Wang, Y.; Feng, S.; Wang, C.; Shao, X.; Lin, W.; Li, B.; et al. Calcineurin Inhibitors Ameliorate PAN -induced Podocyte Injury through the NFAT–Angptl4 Pathway. J. Pathol. 2020, 252, 227–238. [Google Scholar] [CrossRef]
- Watts, A.J.B.; Keller, K.H.; Lerner, G.; Rosales, I.; Collins, A.B.; Sekulic, M.; Waikar, S.S.; Chandraker, A.; Riella, L.V.; Alexander, M.P.; et al. Discovery of Autoantibodies Targeting Nephrin in Minimal Change Disease Supports a Novel Autoimmune Etiology. J. Am. Soc. Nephrol. 2022, 33, 238–252. [Google Scholar] [CrossRef]
- Lin, L.; Hu, K. Annexin A2 and Kidney Diseases. Front. Cell Dev. Biol. 2022, 10, 974381. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhang, Y.; Zhuang, J.; Bi, Y.; Xu, H.; Shen, Q.; Liu, J.; Fu, H.; Wang, J.; Feng, C.; et al. The Important Roles and Molecular Mechanisms of Annexin A2 Autoantibody in Children with Nephrotic Syndrome. Ann. Transl. Med. 2021, 9, 1452. [Google Scholar] [CrossRef]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic Kidney Disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Mohandes, S.; Doke, T.; Hu, H.; Mukhi, D.; Dhillon, P.; Susztak, K. Molecular Pathways That Drive Diabetic Kidney Disease. J. Clin. Investig. 2023, 133, e165654. [Google Scholar] [CrossRef] [PubMed]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Rajendran, M.; Rostovtseva, T.; Hool, L. How Cytoskeletal Proteins Regulate Mitochondrial Energetics in Cell Physiology and Diseases. Phil. Trans. R. Soc. B 2022, 377, 20210324. [Google Scholar] [CrossRef]
- Oosterheert, W.; Klink, B.U.; Belyy, A.; Pospich, S.; Raunser, S. Structural Basis of Actin Filament Assembly and Aging. Nature 2022, 611, 374–379. [Google Scholar] [CrossRef]
- Plastino, J.; Blanchoin, L. Dynamic Stability of the Actin Ecosystem. J. Cell Sci. 2019, 132, jcs219832. [Google Scholar] [CrossRef]
- Wolff, D.W.; Bianchi-Smiraglia, A.; Nikiforov, M.A. Compartmentalization and Regulation of GTP in Control of Cellular Phenotypes. Trends Mol. Med. 2022, 28, 758–769. [Google Scholar] [CrossRef]
- Matsuda, J.; Asano-Matsuda, K.; Kitzler, T.M.; Takano, T. Rho GTPase Regulatory Proteins in Podocytes. Kidney Int. 2021, 99, 336–345. [Google Scholar] [CrossRef]
- Shen, J.; Wang, R.; He, Z.; Huang, H.; He, X.; Zhou, J.; Yan, Y.; Shen, S.; Shao, X.; Shen, X.; et al. NMDA Receptors Participate in the Progression of Diabetic Kidney Disease by Decreasing Cdc42-GTP Activation in Podocytes: NMDARs in DKD Podocytes. J. Pathol. 2016, 240, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Anderson, M.; Dryer, S.E. Insulin Increases Surface Expression of TRPC6 Channels in Podocytes: Role of NADPH Oxidases and Reactive Oxygen Species. Am. J. Physiol.-Ren. Physiol. 2012, 302, F298–F307. [Google Scholar] [CrossRef] [PubMed]
- Rachubik, P.; Szrejder, M.; Rogacka, D.; Audzeyenka, I.; Rychłowski, M.; Angielski, S.; Piwkowska, A. The TRPC6-AMPK Pathway Is Involved in Insulin-Dependent Cytoskeleton Reorganization and Glucose Uptake in Cultured Rat Podocytes. Cell Physiol. Biochem. 2018, 51, 393–410. [Google Scholar] [CrossRef]
- Rachubik, P.; Szrejder, M.; Rogacka, D.; Typiak, M.; Audzeyenka, I.; Kasztan, M.; Pollock, D.M.; Angielski, S.; Piwkowska, A. Insulin Controls Cytoskeleton Reorganization and Filtration Barrier Permeability via the PKGIα-Rac1-RhoA Crosstalk in Cultured Rat Podocytes. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2022, 1869, 119301. [Google Scholar] [CrossRef] [PubMed]
- Piwkowska, A. Role of Protein Kinase G and Reactive Oxygen Species in the Regulation of Podocyte Function in Health and Disease: PODOCYTE AND PKGIα-DEPENDENT SIGNALING. J. Cell. Physiol. 2017, 232, 691–697. [Google Scholar] [CrossRef]
- Audzeyenka, I.; Rogacka, D.; Rachubik, P.; Typiak, M.; Rychłowski, M.; Angielski, S.; Piwkowska, A. The PKGIα–Rac1 Pathway Is a Novel Regulator of Insulin-dependent Glucose Uptake in Cultured Rat Podocytes. J. Cell Physiol. 2021, 236, 4655–4668. [Google Scholar] [CrossRef]
- Armelloni, S.; Calvaresi, N.; Ikehata, M.; Corbelli, A.; Mattinzoli, D.; Giardino, L.A.; Li, M.; Messa, P.; Rastaldi, M.P. Proteinuria and Glomerular Damage in Rab3A Knockout Mice Chronically Fed a High-Glucose Diet. Nephron Exp. Nephrol. 2012, 120, e69–e80. [Google Scholar] [CrossRef]
- Martinez-Arroyo, O.; Flores-Chova, A.; Sanchez-Garcia, B.; Redon, J.; Cortes, R.; Ortega, A. Rab3A/Rab27A System Silencing Ameliorates High Glucose-Induced Injury in Podocytes. Biology 2023, 12, 690. [Google Scholar] [CrossRef]
- Huang, Z.; Peng, Y.; Yu, H.; Yu, X.; Zhou, J.; Xiao, J. RhoA Protects the Podocytes against High Glucose-Induced Apoptosis through YAP and Plays Critical Role in Diabetic Nephropathy. Biochem. Biophys. Res. Commun. 2018, 504, 949–956. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, S.; Shi, J.; Xu, X.; Wang, L.; Zhai, X.; Hou, Q.; Qin, W.; Chen, Z. TYRO3 Protects Podocyte via JNK/c-Jun-P53 Pathway. Arch. Biochem. Biophys. 2023, 739, 109578. [Google Scholar] [CrossRef]
- Guo, K.; Pan, P.; Wu, M.; Ma, Y.; Lu, J.; Chen, H. Hyposialylated Angiopoietin-like-4 Induces Apoptosis of Podocytes via Β1 Integrin/FAK Signaling in Diabetic Nephropathy. Mol. Cell. Endocrinol. 2020, 505, 110730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, C.; Ye, Q.; Tong, L.; Jiang, H.; Zhu, X.; Huang, L.; Lin, W.; Fu, H.; Wang, J.; et al. Podocyte Apoptosis in Diabetic Nephropathy by BASP1 Activation of the P53 Pathway via WT1. Acta Physiol. 2021, 232. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, F.; Rousseau, M.; Denhez, B.; Lévesque, D.; Guay, A.; Liu, H.; Moreau, J.; Higgins, S.; Sabbagh, R.; Susztak, K.; et al. Deletion of Protein Tyrosine Phosphatase SHP-1 Restores SUMOylation of Podocin and Reverses the Progression of Diabetic Kidney Disease. Kidney Int. 2023, 104, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Lee, S.J.; Lee, J.-H.; Kim, J.-H.; Son, S.S.; Cha, S.-K.; Lee, E.S.; Chung, C.H.; Lee, E.Y. Angiotensin II-Mediated MYH9 Downregulation Causes Structural and Functional Podocyte Injury in Diabetic Kidney Disease. Sci. Rep. 2019, 9, 7679. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Hidaka, T.; Nakayama, M.; Sasaki, Y.; Takagi, M.; Suzuki, H.; Suzuki, Y. Protective Effects of DPP-4 Inhibitor on Podocyte Injury in Glomerular Diseases. BMC Nephrol. 2020, 21, 402. [Google Scholar] [CrossRef]
- Pan, Y.; Jiang, S.; Hou, Q.; Qiu, D.; Shi, J.; Wang, L.; Chen, Z.; Zhang, M.; Duan, A.; Qin, W.; et al. Dissection of Glomerular Transcriptional Profile in Patients with Diabetic Nephropathy: SRGAP2a Protects Podocyte Structure and Function. Diabetes 2018, 67, 717–730. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Q.; Fan, X.; Zhen, J.; Wang, C.; Chen, H.; Liu, Y.; Zhou, P.; Zhang, T.; Huang, T.; et al. Long Noncoding RNA ENST00000436340 Promotes Podocyte Injury in Diabetic Kidney Disease by Facilitating the Association of PTBP1 with RAB3B. Cell Death Dis. 2023, 14, 130. [Google Scholar] [CrossRef]
- Garsen, M.; Rops, A.L.W.M.M.; Dijkman, H.; Willemsen, B.; Van Kuppevelt, T.H.; Russel, F.G.; Rabelink, T.J.; Berden, J.H.M.; Reinheckel, T.; Van Der Vlag, J. Cathepsin L Is Crucial for the Development of Early Experimental Diabetic Nephropathy. Kidney Int. 2016, 90, 1012–1022. [Google Scholar] [CrossRef]
- Yaddanapudi, S.; Altintas, M.M.; Kistler, A.D.; Fernandez, I.; Möller, C.C.; Wei, C.; Peev, V.; Flesche, J.B.; Forst, A.-L.; Li, J.; et al. CD2AP in Mouse and Human Podocytes Controls a Proteolytic Program that Regulates Cytoskeletal Structure and Cellular Survival. J. Clin. Investig. 2011, 121, 3965–3980. [Google Scholar] [CrossRef]
- Sever, S.; Altintas, M.M.; Nankoe, S.R.; Möller, C.C.; Ko, D.; Wei, C.; Henderson, J.; Del Re, E.C.; Hsing, L.; Erickson, A.; et al. Proteolytic Processing of Dynamin by Cytoplasmic Cathepsin L Is a Mechanism for Proteinuric Kidney Disease. J. Clin. Investig. 2007, 117, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized Roles for Cysteine Cathepsins in Health and Disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Li, M.; Qiu, Y.; Xie, Y.; Hao, Z.; Yin, X.; Zhang, Z.; Su, H.; Yang, L.; Lin, J.; et al. Asparaginyl Endopeptidase Protects against Podocyte Injury in Diabetic Nephropathy through Cleaving Cofilin-1. Cell Death Dis. 2022, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Beck, L.; Debiec, H.; Fervenza, F.C.; Hou, F.F.; Jha, V.; Sethi, S.; Tong, A.; Vivarelli, M.; Wetzels, J. Membranous Nephropathy. Nat. Rev. Dis. Primers 2021, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Haddad, G.; Lorenzen, J.M.; Ma, H.; De Haan, N.; Seeger, H.; Zaghrini, C.; Brandt, S.; Kölling, M.; Wegmann, U.; Kiss, B.; et al. Altered Glycosylation of IgG4 Promotes Lectin Complement Pathway Activation in Anti-PLA2R1–Associated Membranous Nephropathy. J. Clin. Investig. 2021, 131, e140453. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C.; Meyer, T.N.; Sievert, H.; Hoxha, E.; Sachs, M.; Klupp, E.-M.; Münster, S.; Balabanov, S.; Carrier, L.; Helmchen, U.; et al. Ubiquitin C-Terminal Hydrolase-L1 Activity Induces Polyubiquitin Accumulation in Podocytes and Increases Proteinuria in Rat Membranous Nephropathy. Am. J. Pathol. 2011, 178, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, F.; Sachs, M.; Meyer, T.N.; Sievert, H.; Lindenmeyer, M.T.; Wiech, T.; Cohen, C.D.; Balabanov, S.; Stahl, R.A.K.; Meyer-Schwesinger, C. UCH-L1 Induces Podocyte Hypertrophy in Membranous Nephropathy by Protein Accumulation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 945–958. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, W.; Sun, Y.; Qiao, Y.; Zhang, L.; Zhao, Z.; Lv, S. Wnt/β-Catenin Signaling Mediated-UCH-L1 Expression in Podocytes of Diabetic Nephropathy. Int. J. Mol. Sci. 2016, 17, 1404. [Google Scholar] [CrossRef]
- Fang, Y.; Li, F.; Qi, C.; Mao, X.; Xu, Y.; Zhao, Z.; Wu, H.; Zhang, Z. Plakoglobin Is Involved in Cytoskeletal Rearrangement of Podocytes under the Regulation of UCH-L1. Biochem. Biophys. Res. Commun. 2020, 529, 112–118. [Google Scholar] [CrossRef]
- Peng, L.; Ma, J.; Cui, R.; Chen, X.; Wei, S.-Y.; Wei, Q.-J.; Li, B. The Calcineurin Inhibitor Tacrolimus Reduces Proteinuria in Membranous Nephropathy Accompanied by a Decrease in Angiopoietin-Like-4. PLoS ONE 2014, 9, e106164. [Google Scholar] [CrossRef]
- Ozawa, S.; Matsubayashi, M.; Nanaura, H.; Yanagita, M.; Mori, K.; Asanuma, K.; Kajiwara, N.; Hayashi, K.; Ohashi, H.; Kasahara, M.; et al. Proteolytic Cleavage of Podocin by Matriptase Exacerbates Podocyte Injury. J. Biol. Chem. 2020, 295, 16002–16012. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, P.; Dang, X.; Zhang, X.; Mao, Y.; Chen, X. Lupus Nephritis: New Progress in Diagnosis and Treatment. J. Autoimmun. 2022, 132, 102871. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, X.; Fu, X.; Yang, Y.; Chen, J.; Lin, W. MicroRNA-26a: An Emerging Regulator of Renal Biology and Disease. Kidney Blood Press. Res. 2019, 44, 287–297. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka-Kanazawa, S.; Horino, T.; Kimura, J.; Nakamura, T.; Matsumoto, M.; Toi, M.; Kon, Y. Decreased miR-26a Expression Correlates with the Progression of Podocyte Injury in Autoimmune Glomerulonephritis. PLoS ONE 2014, 9, e110383. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Caster, D.J.; Korte, E.A.; Tan, M.; Barati, M.T.; Tandon, S.; Creed, T.M.; Salant, D.J.; Hata, J.L.; Epstein, P.N.; Huang, H.; et al. Neutrophil Exocytosis Induces Podocyte Cytoskeletal Reorganization and Proteinuria in Experimental Glomerulonephritis. Am. J. Physiol.-Ren. Physiol. 2018, 315, F595–F606. [Google Scholar] [CrossRef]
- Mathieson, P.W. The Podocyte as a Target for Therapies—New and Old. Nat. Rev. Nephrol. 2012, 8, 52–56. [Google Scholar] [CrossRef]
- Lee, H.W.; Arif, E.; Altintas, M.M.; Quick, K.; Maheshwari, S.; Plezia, A.; Mahmood, A.; Reiser, J.; Nihalani, D.; Gupta, V. High-Content Screening Assay-Based Discovery of Paullones as Novel Podocyte-Protective Agents. Am. J. Physiol.-Ren. Physiol. 2018, 314, F280–F292. [Google Scholar] [CrossRef]
- Ponticelli, C.; Locatelli, F. Glucocorticoids in the Treatment of Glomerular Diseases: Pitfalls and Pearls. Clin. J. Am. Soc. Nephrol. 2018, 13, 815–822. [Google Scholar] [CrossRef]
- Wang, H.; Duan, A.; Zhang, J.; Wang, Q.; Xing, Y.; Qin, Z.; Liu, Z.; Yang, J. Glucocorticoid Receptor Wields Chromatin Interactions to Tune Transcription for Cytoskeleton Stabilization in Podocytes. Commun. Biol. 2021, 4, 675. [Google Scholar] [CrossRef]
- van den Broek, M.; Smeets, B.; Schreuder, M.F.; Jansen, J. The Podocyte as a Direct Target of Glucocorticoids in Nephrotic Syndrome. Nephrol. Dial. Transplant. 2022, 37, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.-Y.; Saleem, M.A.; Coward, R.J.; Ni, L.; Witherden, I.R.; Mathieson, P.W. Direct Effects of Dexamethasone on Human Podocytes. Kidney Int. 2006, 70, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Khoshnoodi, J.; Takenaka, H.; Hosoyamada, M.; Nakajo, A.; Bessho, F.; Kudo, A.; Takahashi, S.; Arimura, Y.; Yamada, A.; et al. The Effect of Dexamethasone on Defective Nephrin Transport Caused by ER Stress: A Potential Mechanism for the Therapeutic Action of Glucocorticoids in the Acquired Glomerular Diseases. Kidney Int. 2006, 69, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Ding, J.; Fan, Q.; Guan, N. Diversities of Podocyte Molecular Changes Induced by Different Antiproteinuria Drugs. Exp. Biol. Med. 2006, 231, 585–593. [Google Scholar] [CrossRef]
- Moysiadis, D.; Perysinaki, G.; Bertsias, G.; Stratakis, S.; Kyriacou, K.; Nakopoulou, L.; Boumpas, D.; Daphnis, E. Early Treatment with Glucocorticoids or Cyclophosphamide Retains the Slit Diaphragm Proteins Nephrin and Podocin in Experimental Lupus Nephritis. Lupus 2012, 21, 1196–1207. [Google Scholar] [CrossRef]
- Yu, M.; Ren, Q.; Yu, S.Y. Role of Nephrin Phosphorylation Inducted by Dexamethasone and Angiotensin II in Podocytes. Mol. Biol. Rep. 2014, 41, 3591–3595. [Google Scholar] [CrossRef]
- Jiang, L.; Hindmarch, C.C.T.; Rogers, M.; Campbell, C.; Waterfall, C.; Coghill, J.; Mathieson, P.W.; Welsh, G.I. RNA Sequencing Analysis of Human Podocytes Reveals Glucocorticoid Regulated Gene Networks Targeting Non-Immune Pathways. Sci. Rep. 2016, 6, 35671. [Google Scholar] [CrossRef]
- Yu, S.; Yu, L. Dexamethasone Resisted Podocyte Injury via Stabilizing TRPC6 Expression and Distribution. Evid.-Based Complement. Altern. Med. 2012, 2012, 652059. [Google Scholar] [CrossRef]
- Liu, H.; Gao, X.; Xu, H.; Feng, C.; Kuang, X.; Li, Z.; Zha, X. α-Actinin-4 Is Involved in the Process by which Dexamethasone Protects Actin Cytoskeleton Stabilization from Adriamycin-Induced Podocyte Injury: Dexamethasone and Cytoskeleton. Nephrology 2012, 17, 669–675. [Google Scholar] [CrossRef]
- McCaffrey, J.C.; Webb, N.J.; Poolman, T.M.; Fresquet, M.; Moxey, C.; Zeef, L.A.H.; Donaldson, I.J.; Ray, D.W.; Lennon, R. Glucocorticoid Therapy Regulates Podocyte Motility by Inhibition of Rac1. Sci. Rep. 2017, 7, 6725. [Google Scholar] [CrossRef]
- Hirakawa, M.; Tsuruya, K.; Yotsueda, H.; Tokumoto, M.; Ikeda, H.; Katafuchi, R.; Fujimi, S.; Hirakata, H.; Iida, M. Expression of Synaptopodin and GLEPP1 as Markers of Steroid Responsiveness in Primary Focal Segmental Glomerulosclerosis. Life Sci. 2006, 79, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Chanley, M.A.; Westbrook, D.; Nie, X.; Kitao, T.; Guess, A.J.; Benndorf, R.; Hidalgo, G.; Smoyer, W.E. Pioglitazone Enhances the Beneficial Effects of Glucocorticoids in Experimental Nephrotic Syndrome. Sci. Rep. 2016, 6, 24392. [Google Scholar] [CrossRef] [PubMed]
- Lewko, B.; Waszkiewicz, A.; Maryn, A.; Gołos, M.; Latawiec, E.; Daca, A.; Witkowski, J.M.; Angielski, S.; Stępiński, J. Dexamethasone-Dependent Modulation of Cyclic GMP Synthesis in Podocytes. Mol. Cell Biochem. 2015, 409, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Guo, Y.; Revelo, M.P.; Roa-Peña, L.; Miller, T.; Ling, J.; Shankland, S.J.; Bialkowska, A.B.; Ly, V.; Estrada, C.; et al. Krüppel–Like Factor 15 Mediates Glucocorticoid-Induced Restoration of Podocyte Differentiation Markers. J. Am. Soc. Nephrol. 2017, 28, 166–184. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pace, J.; Li, Z.; Ma’ayan, A.; Wang, Z.; Revelo, M.P.; Chen, E.; Gu, X.; Attalah, A.; Yang, Y.; et al. Podocyte-Specific Induction of Krüppel-Like Factor 15 Restores Differentiation Markers and Attenuates Kidney Injury in Proteinuric Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2529–2545. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Zhang, H.; Ke, G.; Zhang, L.; Lian, Z.; Chen, X.; Zhao, X.; Chen, Y.; Li, R.; Ma, J.; et al. The Krüppel-like Factor 15-NFATc1 Axis Ameliorates Podocyte Injury: A Novel Rationale for Using Glucocorticoids in Proteinuria Diseases. Clin. Sci. 2020, 134, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Bharati, J.; Nada, R.; Minz, R.; Kohli, H.S. Rituximab in Maintaining Remission in Adults with Podocytopathy. Nephrology 2020, 25, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, R.; Baldovino, S.; Sciascia, S.; De Simone, E.; Del Vecchio, G.; Ferro, M.; Quattrocchio, G.; Naretto, C.; Roccatello, D. Efficacy of Low or Standard Rituximab-Based Protocols and Comparison to Ponticelli’s Regimen in Membranous Nephropathy. J. Nephrol. 2021, 34, 565–571. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ikezumi, Y.; Saitoh, A. Rituximab Protects Podocytes and Exerts Anti-Proteinuric Effects in Rat Adriamycin-Induced Nephropathy Independent of B-Lymphocytes: Rituximab Ameliorates Proteinuria in Rat ADN. Nephrology 2017, 22, 49–57. [Google Scholar] [CrossRef]
- Jeruschke, S.; Alex, D.; Hoyer, P.F.; Weber, S. Protective Effects of Rituximab on Puromycin-Induced Apoptosis, Loss of Adhesion and Cytoskeletal Alterations in Human Podocytes. Sci. Rep. 2022, 12, 12297. [Google Scholar] [CrossRef]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. Generation of Biologically Active Linear and Cyclic Peptides Has Revealed a Unique Fine Specificity of Rituximab and Its Possible Cross-Reactivity with Acid Sphingomyelinase-like Phosphodiesterase 3b Precursor. Blood 2006, 107, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab Targets Podocytes in Recurrent Focal Segmental Glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Book, B.K.; Sidner, R.A. Resolution of Recurrent Focal Segmental Glomerulosclerosis Proteinuria after Rituximab Treatment. N. Engl. J. Med. 2006, 354, 1961–1963. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Faguer, S.; Esposito, L.; Guitard, J.; Nogier, M.B.; Durand, D.; Rostaing, L. Treatment of Focal Segmental Glomerular Sclerosis with Rituximab: 2 Case Reports. Clin. Nephrol. 2007, 67, 250–254. [Google Scholar] [CrossRef]
- Strologo, L.D.; Guzzo, I.; Laurenzi, C.; Vivarelli, M.; Parodi, A.; Barbano, G.; Camilla, R.; Scozzola, F.; Amore, A.; Ginevri, F.; et al. Use of Rituximab in Focal Glomerulosclerosis Relapses after Renal Transplantation. Transplantation 2009, 88, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Takasu, J.; Nihei, H.; Yonekura, T.; Aoki, Y.; Kawamura, T.; Mizuiri, S.; Aikawa, A. Protocol Biopsies for Focal Segmental Glomerulosclerosis Treated with Plasma Exchange and Rituximab in a Renal Transplant Patient: Plasma Exchange and Rituximab for FSGS. Clin. Transplant. 2010, 24, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Reggiani, F.; Moroni, G. Old and New Calcineurin Inhibitors in Lupus Nephritis. J. Clin. Med. 2021, 10, 4832. [Google Scholar] [CrossRef]
- Clipstone, N.A.; Crabtree, G.R. Identification of Calcineurin as a Key Signalling Enzyme in T-Lymphocyte Activation. Nature 1992, 357, 695–697. [Google Scholar] [CrossRef]
- Descazeaud, V.; Mestre, E.; Marquet, P.; Essig, M. Calcineurin Regulation of Cytoskeleton Organization: A New Paradigm to Analyse the Effects of Calcineurin Inhibitors on the Kidney. J. Cell. Mol. Med. 2012, 16, 218–227. [Google Scholar] [CrossRef]
- Mundel, P.; Reiser, J. Proteinuria: An Enzymatic Disease of the Podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.-M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The Actin Cytoskeleton of Kidney Podocytes Is a Direct Target of the Antiproteinuric Effect of Cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Liu, Q.; Zheng, Z.; Fan, J.; Peng, W.; Kong, Q.; He, H.; Yang, S.; Chen, W.; Tang, X.; et al. Tacrolimus Protects Podocytes from Injury in Lupus Nephritis Partly by Stabilizing the Cytoskeleton and Inhibiting Podocyte Apoptosis. PLoS ONE 2015, 10, e0132724. [Google Scholar] [CrossRef]
- Sever, S.; Schiffer, M. Actin Dynamics at Focal Adhesions: A Common Endpoint and Putative Therapeutic Target for Proteinuric Kidney Diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, T.; Sloan, A.J.; Hoenderop, J.G.J.; Flesche, J.; Van Goor, H.; Kistler, A.D.; Bakker, M.; Bindels, R.J.M.; De Boer, R.A.; Möller, C.C.; et al. Angiotensin II Contributes to Podocyte Injury by Increasing TRPC6 Expression via an NFAT-Mediated Positive Feedback Signaling Pathway. Am. J. Pathol. 2011, 179, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Schlöndorff, J.; Del Camino, D.; Carrasquillo, R.; Lacey, V.; Pollak, M.R. TRPC6 Mutations Associated with Focal Segmental Glomerulosclerosis Cause Constitutive Activation of NFAT-Dependent Transcription. Am. J. Physiol.-Cell Physiol. 2009, 296, C558–C569. [Google Scholar] [CrossRef] [PubMed]
- Shengyou, Y.; Li, Y.; Zhihong, H.; Yuanyuan, M. Influence of Tacrolimus on Podocyte Injury Inducted by Angiotensin II. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, Y.; Li, X.; Thilo, F.; Tepel, M. Insulin Increases Expression of TRPC6 Channels in Podocytes by a Calcineurin-Dependent Pathway. Cell Physiol. Biochem. 2016, 38, 659–669. [Google Scholar] [CrossRef]
- Li, X.; Ding, F.; Wang, S.; Li, B.; Ding, J. Cyclosporine A Protects Podocytes by Regulating WAVE1 Phosphorylation. Sci. Rep. 2015, 5, 17694. [Google Scholar] [CrossRef]
- Shen, X.; Weng, C.; Wang, Y.; Wang, C.; Feng, S.; Li, X.; Li, H.; Jiang, H.; Wang, H.; Chen, J. Lipopolysaccharide-Induced Podocyte Injury Is Regulated by Calcineurin/NFAT and TLR4/MyD88/NF-κB Signaling Pathways through Angiopoietin-like Protein 4. Genes Dis. 2022, 9, 443–455. [Google Scholar] [CrossRef]
- Li, J.-S.; Chen, X.; Peng, L.; Wei, S.-Y.; Zhao, S.-L.; Diao, T.-T.; He, Y.-X.; Liu, F.; Wei, Q.-J.; Zhang, Q.-F.; et al. Angiopoietin-Like-4, a Potential Target of Tacrolimus, Predicts Earlier Podocyte Injury in Minimal Change Disease. PLoS ONE 2015, 10, e0137049. [Google Scholar] [CrossRef]
- Yasuda, H.; Fukusumi, Y.; Ivanov, V.; Zhang, Y.; Kawachi, H. Tacrolimus Ameliorates Podocyte Injury by Restoring FK506 Binding Protein 12 (FKBP12) at Actin Cytoskeleton. FASEB J. 2021, 35, e21983. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-M.; Wang, J.; Xu, X.-X.; Li, Y.-Y.; Wu, Y.-G. FK506 Reduces Albuminuria through Improving Podocyte Nephrin and Podocin Expression in Diabetic Rats. Inflamm. Res. 2016, 65, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, S.; Yu, L.; Ge, L.; Zhang, Y.; Hao, Z.; Liu, G. Effects of Tacrolimus on Autophagy Protein LC3 in Puromycin-Damaged Mouse Podocytes. J. Int. Med. Res. 2020, 48, 300060520971422. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Dong, W.; Chen, Y.; Tang, T.; Zhao, X.; Zhang, L.; Liang, X. Effect of Cyclosporine A on Focal Segmental Glomerulosclerosis Caused by MYO1E Mutation in a Chinese Adult Patient: A Case Report. Medicine 2023, 102, e32683. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, I.; Terai, C.; Yabe, H.; Sugawara, H. Tacrolimus Induction Therapy for Nephrotic Syndrome Caused by Minimal Mesangial Lupus Nephritis with Lupus Podocytopathy: A Case-Based Review. Am. J. Case Rep. 2022, 23, e937201. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Sharpe, A.H. T-Cell Stimulation: An Abundance of B7s. Nat. Med. 1999, 5, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in Podocytes Is Associated with Nephrotic Syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-C.; Fornoni, A.; Weins, A.; Hakroush, S.; Maiguel, D.; Sageshima, J.; Chen, L.; Ciancio, G.; Faridi, M.H.; Behr, D.; et al. Abatacept in B7-1–Positive Proteinuric Kidney Disease. N. Engl. J. Med. 2013, 369, 2416–2423. [Google Scholar] [CrossRef]
- Genovese, M.C.; Becker, J.-C.; Schiff, M.; Luggen, M.; Sherrer, Y.; Kremer, J.; Birbara, C.; Box, J.; Natarajan, K.; Nuamah, I.; et al. Abatacept for Rheumatoid Arthritis Refractory to Tumor Necrosis Factor α Inhibition. N. Engl. J. Med. 2005, 353, 1114–1123. [Google Scholar] [CrossRef]
- Ruperto, N.; Lovell, D.J.; Quartier, P.; Paz, E.; Rubio-Pérez, N.; Silva, C.A.; Abud-Mendoza, C.; Burgos-Vargas, R.; Gerloni, V.; Melo-Gomes, J.A.; et al. Abatacept in Children with Juvenile Idiopathic Arthritis: A Randomised, Double-Blind, Placebo-Controlled Withdrawal Trial. Lancet 2008, 372, 383–391. [Google Scholar] [CrossRef]
- Delville, M.; Baye, E.; Durrbach, A.; Audard, V.; Kofman, T.; Braun, L.; Olagne, J.; Nguyen, C.; Deschênes, G.; Moulin, B.; et al. B7–1 Blockade Does Not Improve Post–Transplant Nephrotic Syndrome Caused by Recurrent FSGS. J. Am. Soc. Nephrol. 2016, 27, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Isom, R.; Shoor, S.; Higgins, J.; Cara-Fuentes, G.; Johnson, R.J. Abatacept in Steroid-Dependent Minimal Change Disease and CD80-Uria. Kidney Int. Rep. 2019, 4, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Mühlbacher, T.; Amann, K.; Mahling, M.; Nadalin, S.; Heyne, N.; Guthoff, M. Successful Long-Term Management of Recurrent Focal Segmental Glomerulosclerosis after Kidney Transplantation with Costimulation Blockade. Clin. Kidney J. 2021, 14, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.W.; Chandar, J.; Sageshima, J.; Ortigosa-Goggins, M.; Amarapurkar, P.; Mitrofanova, A.; Defreitas, M.J.; Katsoufis, C.P.; Seeherunvong, W.; Centeno, A.; et al. Benefit of B7-1 Staining and Abatacept for Treatment-Resistant Post-Transplant Focal Segmental Glomerulosclerosis in a Predominantly Pediatric Cohort: Time for a Reappraisal. Pediatr. Nephrol. 2023, 38, 145–159. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT02592798?cond=NCT02592798&draw=2&rank=1 (accessed on 8 July 2023).
- Fiorina, P.; Vergani, A.; Bassi, R.; Niewczas, M.A.; Altintas, M.M.; Pezzolesi, M.G.; D’Addio, F.; Chin, M.; Tezza, S.; Ben Nasr, M.; et al. Role of Podocyte B7-1 in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1415–1429. [Google Scholar] [CrossRef]
- Gagliardini, E.; Novelli, R.; Corna, D.; Zoja, C.; Ruggiero, B.; Benigni, A.; Remuzzi, G. B7–1 Is not Induced in Podocytes of Human and Experimental Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 999–1005. [Google Scholar] [CrossRef][Green Version]
- Herrera, M.; Söderberg, M.; Sabirsh, A.; Valastro, B.; Mölne, J.; Santamaria, B.; Valverde, A.M.; Guionaud, S.; Heasman, S.; Bigley, A.; et al. Inhibition of T-Cell Activation by the CTLA4-Fc Abatacept Is Sufficient to Ameliorate Proteinuric Kidney Disease. Am. J. Physiol.-Ren. Physiol. 2017, 312, F748–F759. [Google Scholar] [CrossRef]
- Norlin, J.; Nielsen Fink, L.; Helding Kvist, P.; Douglas Galsgaard, E.; Coppieters, K. Abatacept Treatment Does Not Preserve Renal Function in the Streptozocin-Induced Model of Diabetic Nephropathy. PLoS ONE 2016, 11, e0152315. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, Y.; Wu, L.; Chen, S.; Han, F. Efficacy and Safety of Biologic Agents for Lupus Nephritis: A Systematic Review and Meta-Analysis. J. Clin. Rheumatol. 2023, 29, 95–100. [Google Scholar] [CrossRef]
- Furie, R.; Nicholls, K.; Cheng, T.; Houssiau, F.; Burgos-Vargas, R.; Chen, S.; Hillson, J.L.; Meadows-Shropshire, S.; Kinaszczuk, M.; Merrill, J.T. Efficacy and Safety of Abatacept in Lupus Nephritis: A Twelve-Month, Randomized, Double-Blind Study. Arthritis Rheumatol. 2014, 66, 379–389. [Google Scholar] [CrossRef]
- The ACCESS Trial Group. Treatment of Lupus Nephritis with Abatacept: The Abatacept and Cyclophosphamide Combination Efficacy and Safety Study: Abatacept in Lupus Nephritis. Arthritis Rheumatol. 2014, 66, 3096–3104. [Google Scholar] [CrossRef]
- Zhang, R.; Lee, D.M.; Jimah, J.R.; Gerassimov, N.; Yang, C.; Kim, S.; Luvsanjav, D.; Winkelman, J.; Mettlen, M.; Abrams, M.E.; et al. Dynamin Regulates the Dynamics and Mechanical Strength of the Actin Cytoskeleton as a Multifilament Actin-Bundling Protein. Nat. Cell Biol. 2020, 22, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Chang, J.; Shchedrina, V.A.; Pham, V.A.; Hartwig, J.H.; Suphamungmee, W.; Lehman, W.; Hyman, B.T.; Bacskai, B.J.; Sever, S. Regulation of Dynamin Oligomerization in Cells: The Role of Dynamin-Actin Interactions and Its GTPase Activity. Traffic 2014, 15, 819–838. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Lee, H.W.; Garborcauskas, G.; Reiser, J.; Gupta, V.; Sever, S. Dynamin Autonomously Regulates Podocyte Focal Adhesion Maturation. J. Am. Soc. Nephrol. 2017, 28, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, K.; Gu, C.; Collins, A.; Mettlen, M.; Samelko, B.; Altintas, M.M.; Sudhini, Y.R.; Wang, X.; Bouley, R.; Brown, D.; et al. Simultaneous Stabilization of Actin Cytoskeleton in Multiple Nephron-Specific Cells Protects the Kidney from Diverse Injury. Nat. Commun. 2022, 13, 2422. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Kume, S.; Yasuda-Yamahara, M.; Yamahara, K.; Takeda, N.; Chin-Kanasaki, M.; Araki, H.; Sekine, O.; Yokoi, H.; Mukoyama, M.; et al. O-Linked β-N-Acetylglucosamine Modification of Proteins Is Essential for Foot Process Maturation and Survival in Podocytes. Nephrol. Dial. Transplant. 2017, 32, 1477–1487. [Google Scholar] [CrossRef]
- Müller-Deile, J.; Teng, B.; Schenk, H.; Haller, H.; Reiser, J.; Sever, S.; Schiffer, M. Drugs Targeting Dynamin Can Restore Cytoskeleton and Focal Contact Alterations of Urinary Podocytes Derived from Patients with Nephrotic Syndrome. Ann. Transl. Med. 2016, 4, 439. [Google Scholar] [CrossRef]
- Kushiyama, T.; Oda, T.; Yamamoto, K.; Higashi, K.; Watanabe, A.; Takechi, H.; Uchida, T.; Oshima, N.; Sakurai, Y.; Miura, S.; et al. Protective Effects of Rho Kinase Inhibitor Fasudil on Rats with Chronic Kidney Disease. Am. J. Physiol.-Ren. Physiol. 2013, 304, F1325–F1334. [Google Scholar] [CrossRef]
- Shibata, S.; Nagase, M.; Fujita, T. Fluvastatin Ameliorates Podocyte Injury in Proteinuric Rats via Modulation of Excessive Rho Signaling. J. Am. Soc. Nephrol. 2006, 17, 754–764. [Google Scholar] [CrossRef]
- Tian, F.; Huang, S.; Xu, W.; Xie, G.; Gan, Y.; Huang, F.; Fan, Y.; Bao, J. Fasudil Compensates Podocyte Injury via CaMK4/Rho GTPases Signal and Actin Cytoskeleton-Dependent Activation of YAP in MRL/Lpr Mice. Int. Immunopharmacol. 2023, 119, 110199. [Google Scholar] [CrossRef]
- Wang, S.; Chen, C.; Su, K.; Zha, D.; Liang, W.; Hillebrands, J.; van Goor, H.; Ding, G. Angiotensin II Induces Reorganization of the Actin Cytoskeleton and Myosin Light-Chain Phosphorylation in Podocytes through Rho/ROCK-Signaling Pathway. Ren. Fail. 2016, 38, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-Y.; Li, C.-Y.; Chen, J.; Pan, L.; Saito, S.; Terashita, T.; Saito, K.; Miyawaki, K.; Shigemoto, K.; Mominoki, K.; et al. Rho-ROCK Signal Pathway Regulates Microtubule-Based Process Formation of Cultured Podocytes—Inhibition of ROCK Promoted Process Elongation. Nephron Exp. Nephrol. 2004, 97, e49–e61. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Fukusumi, Y.; Yamazaki, M.; Kayaba, M.; Kitazawa, Y.; Tomita, M.; Kawachi, H. Angiotensin II Type 1 Receptor Blockade Ameliorates Proteinuria in Puromycin Aminonucleoside Nephropathy by Inhibiting the Reduction of NEPH1 and Nephrin. J. Nephrol. 2014, 27, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, W.; Zhang, Y.; Cheng, Y.; Xu, Z. Effect of Angiotensin II Type 1 Receptor Blocker on 12-Lipoxygenase Activity and Slit Diaphragm Protein Expression in Type 2 Diabetic Rat Glomeruli. J. Nephrol. 2016, 29, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Eckel, J.; Lavin, P.J.; Finch, E.A.; Mukerji, N.; Burch, J.; Gbadegesin, R.; Wu, G.; Bowling, B.; Byrd, A.; Hall, G.; et al. TRPC6 Enhances Angiotensin II-Induced Albuminuria. J. Am. Soc. Nephrol. 2011, 22, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II Has Acute Effects on TRPC6 Channels in Podocytes of Freshly Isolated Glomeruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Jeruschke, S.; Büscher, A.K.; Oh, J.; Saleem, M.A.; Hoyer, P.F.; Weber, S.; Nalbant, P. Protective Effects of the mTOR Inhibitor Everolimus on Cytoskeletal Injury in Human Podocytes Are Mediated by RhoA Signaling. PLoS ONE 2013, 8, e55980. [Google Scholar] [CrossRef] [PubMed]
- Jeruschke, S.; Jeruschke, K.; DiStasio, A.; Karaterzi, S.; Büscher, A.K.; Nalbant, P.; Klein-Hitpass, L.; Hoyer, P.F.; Weiss, J.; Stottmann, R.W.; et al. Everolimus Stabilizes Podocyte Microtubules via Enhancing TUBB2B and DCDC2 Expression. PLoS ONE 2015, 10, e0137043. [Google Scholar] [CrossRef]
- Bergwall, L.; Wallentin, H.; Elvin, J.; Liu, P.; Boi, R.; Sihlbom, C.; Hayes, K.; Wright, D.; Haraldsson, B.; Nyström, J.; et al. Amplification of the Melanocortin-1 Receptor in Nephrotic Syndrome Identifies a Target for Podocyte Cytoskeleton Stabilization. Sci. Rep. 2018, 8, 15731. [Google Scholar] [CrossRef]
- Chen, B.; Alam, Z.; Ge, Y.; Dworkin, L.; Gong, R. Pharmacological Melanocortin 5 Receptor Activation Attenuates Glomerular Injury and Proteinuria in Rats With Puromycin Aminonucleoside Nephrosis. Front. Physiol. 2022, 13, 887641. [Google Scholar] [CrossRef]
- Elvin, J.; Buvall, L.; Lindskog Jonsson, A.; Granqvist, A.; Lassén, E.; Bergwall, L.; Nyström, J.; Haraldsson, B. Melanocortin 1 Receptor Agonist Protects Podocytes through Catalase and RhoA Activation. Am. J. Physiol.-Ren. Physiol. 2016, 310, F846–F856. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, P.; Chang, M.; Chen, B.; Ge, Y.; Malhotra, D.K.; Dworkin, L.D.; Gong, R. Melanocortin Therapy Ameliorates Podocytopathy and Proteinuria in Experimental Focal Segmental Glomerulosclerosis Involving a Podocyte Specific Non-MC1R-Mediated Melanocortinergic Signaling. Clin. Sci. 2020, 134, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.-C.; Miyata, K.N.; Chang, S.-Y.; Zhao, X.-P.; Lo, C.-S.; El-Mortada, M.-A.; Peng, J.; Chenier, I.; Yamashita, M.; Ingelfinger, J.R.; et al. Angiotensin II Type-2-Receptor Stimulation Ameliorates Focal and Segmental Glomerulosclerosis in Mice. Clin. Sci. 2022, 136, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, X.; Zhai, X.; Wang, G.; Qin, W.; Cheng, Z.; Chen, Z. CDDO-Me Ameliorates Podocyte Injury through Anti-Oxidative Stress and Regulation of Actin Cytoskeleton in Adriamycin Nephropathy. Biomed. Pharmacother. 2023, 167, 115617. [Google Scholar] [CrossRef] [PubMed]
- Benetti, A.; Martins, F.L.; Sene, L.B.; Shimizu, M.H.M.; Seguro, A.C.; Luchi, W.M.; Girardi, A.C.C. Urinary DPP4 Correlates with Renal Dysfunction, and DPP4 Inhibition Protects against the Reduction in Megalin and Podocin Expression in Experimental CKD. Am. J. Physiol.-Ren. Physiol. 2021, 320, F285–F296. [Google Scholar] [CrossRef] [PubMed]
- Szrejder, M.; Rachubik, P.; Rogacka, D.; Audzeyenka, I.; Rychłowski, M.; Kreft, E.; Angielski, S.; Piwkowska, A. Metformin Reduces TRPC6 Expression through AMPK Activation and Modulates Cytoskeleton Dynamics in Podocytes under Diabetic Conditions. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165610. [Google Scholar] [CrossRef] [PubMed]
- Suk Kang, J.; Son, S.S.; Lee, J.-H.; Lee, S.W.; Jeong, A.R.; Lee, E.S.; Cha, S.-K.; Chung, C.H.; Lee, E.Y. Protective Effects of Klotho on Palmitate-Induced Podocyte Injury in Diabetic Nephropathy. PLoS ONE 2021, 16, e0250666. [Google Scholar] [CrossRef]
- Yao, X.; Guo, H.; Sun, M.; Meng, S.; Zhu, B.; Fang, J.; Huang, J.; Wang, H.; Xing, L. Klotho Ameliorates Podocyte Injury through Targeting TRPC6 Channel in Diabetic Nephropathy. J. Diabetes Res. 2022, 2022, 1329380. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Shan, Z.; Mi, W.; Zhao, Y.; Li, M.; Wang, B.; Zheng, X.; Feng, W. Catalpol Ameliorates Podocyte Injury by Stabilizing Cytoskeleton and Enhancing Autophagy in Diabetic Nephropathy. Front. Pharmacol. 2019, 10, 1477. [Google Scholar] [CrossRef]
- Liu, J.; Sun, M.; Xia, Y.; Cui, X.; Jiang, J. Phloretin Ameliorates Diabetic Nephropathy by Inhibiting Nephrin and Podocin Reduction through a Non-Hypoglycemic Effect. Food Funct. 2022, 13, 6613–6622. [Google Scholar] [CrossRef]
- Wei, L.; Yong, J.; Zhang, X.; Ling, C.; Wu, Y.; Xu, Z.; Zhang, H.; Cao, X.; Sheng, L.; Zhang, Q.; et al. Shenqi Granule Upregulates CD2AP and α-Actinin4 and Activates Autophagy through Regulation of mTOR/ULK1 Pathway in MPC5 Cells. J. Ethnopharmacol. 2023, 303, 115942. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.L.; Rood, I.M.; Deegens, J.K.J.; Klein, J.B. Isolation and Characterization of Urinary Extracellular Vesicles: Implications for Biomarker Discovery. Nat. Rev. Nephrol. 2017, 13, 731–749. [Google Scholar] [CrossRef] [PubMed]
- Thongboonkerd, V. Roles for Exosome in Various Kidney Diseases and Disorders. Front. Pharmacol. 2020, 10, 1655. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Qiu, Y.; Zhang, C. Cytoskeleton Rearrangement in Podocytopathies: An Update. Int. J. Mol. Sci. 2024, 25, 647. https://doi.org/10.3390/ijms25010647
Ma S, Qiu Y, Zhang C. Cytoskeleton Rearrangement in Podocytopathies: An Update. International Journal of Molecular Sciences. 2024; 25(1):647. https://doi.org/10.3390/ijms25010647
Chicago/Turabian StyleMa, Sijia, Yang Qiu, and Chun Zhang. 2024. "Cytoskeleton Rearrangement in Podocytopathies: An Update" International Journal of Molecular Sciences 25, no. 1: 647. https://doi.org/10.3390/ijms25010647
APA StyleMa, S., Qiu, Y., & Zhang, C. (2024). Cytoskeleton Rearrangement in Podocytopathies: An Update. International Journal of Molecular Sciences, 25(1), 647. https://doi.org/10.3390/ijms25010647