
Endurance Exercise Training Mitigates Diastolic Dysfunction in Diabetic Mice Independent of Phosphorylation of Ulk1 at S555

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
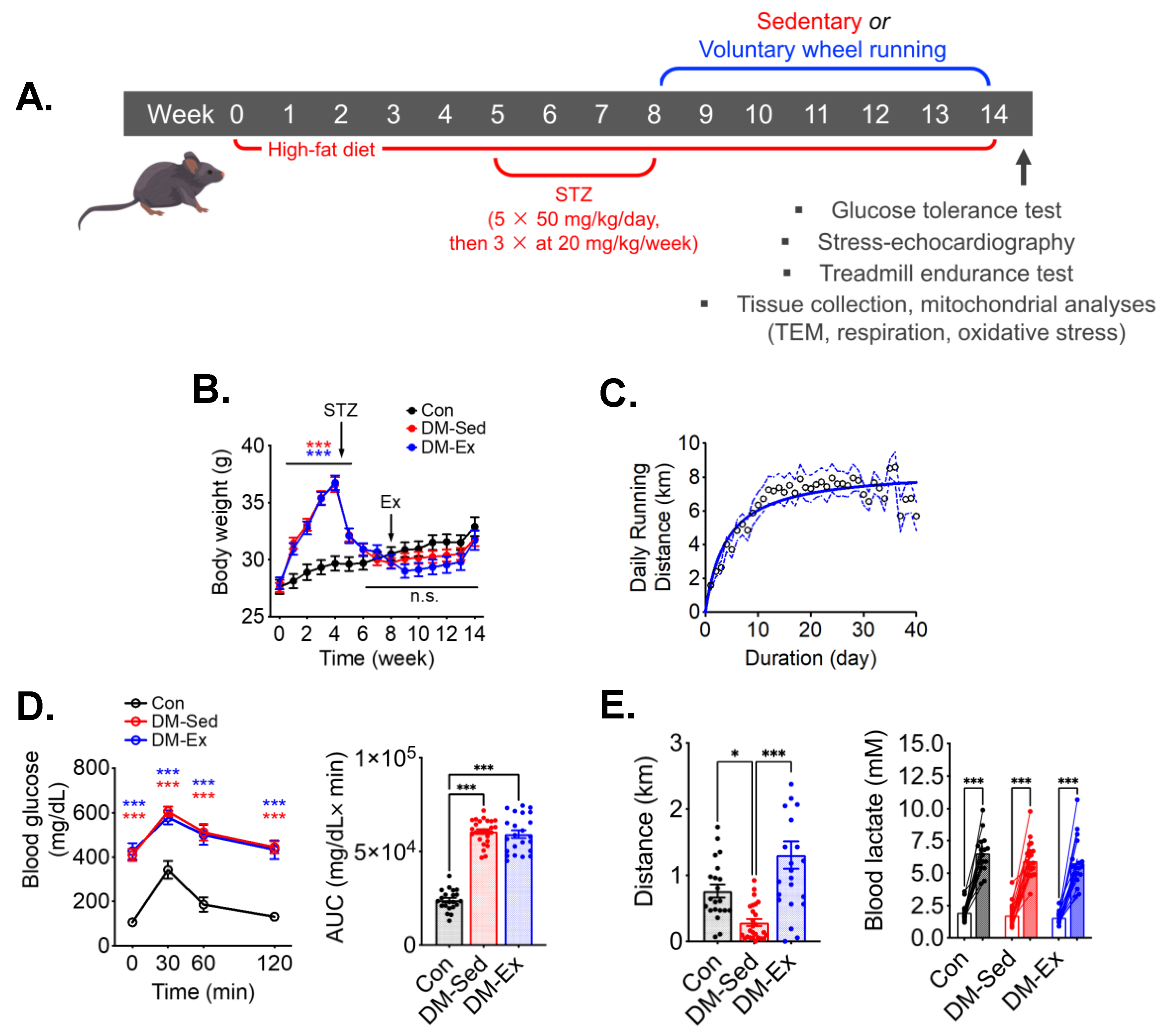
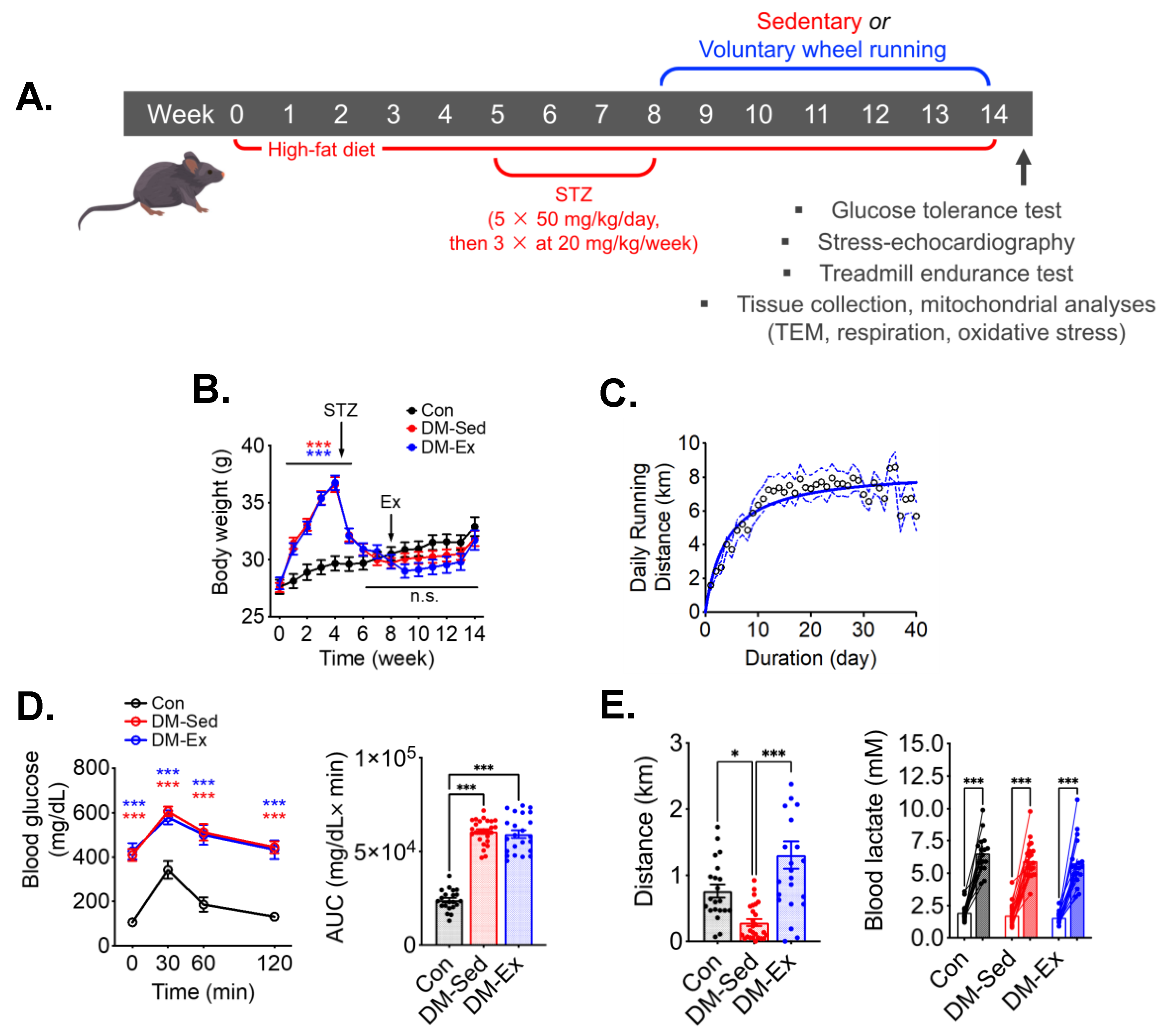
2.1. A Combination of High-Fat Diet (HFD) and Streptozotocin (STZ) Injections Induces Severe Diabetes in Mice
2.2. Diabetic Mice Develop Exercise Intolerance, Which Is Mitigated by Voluntary Wheel Running
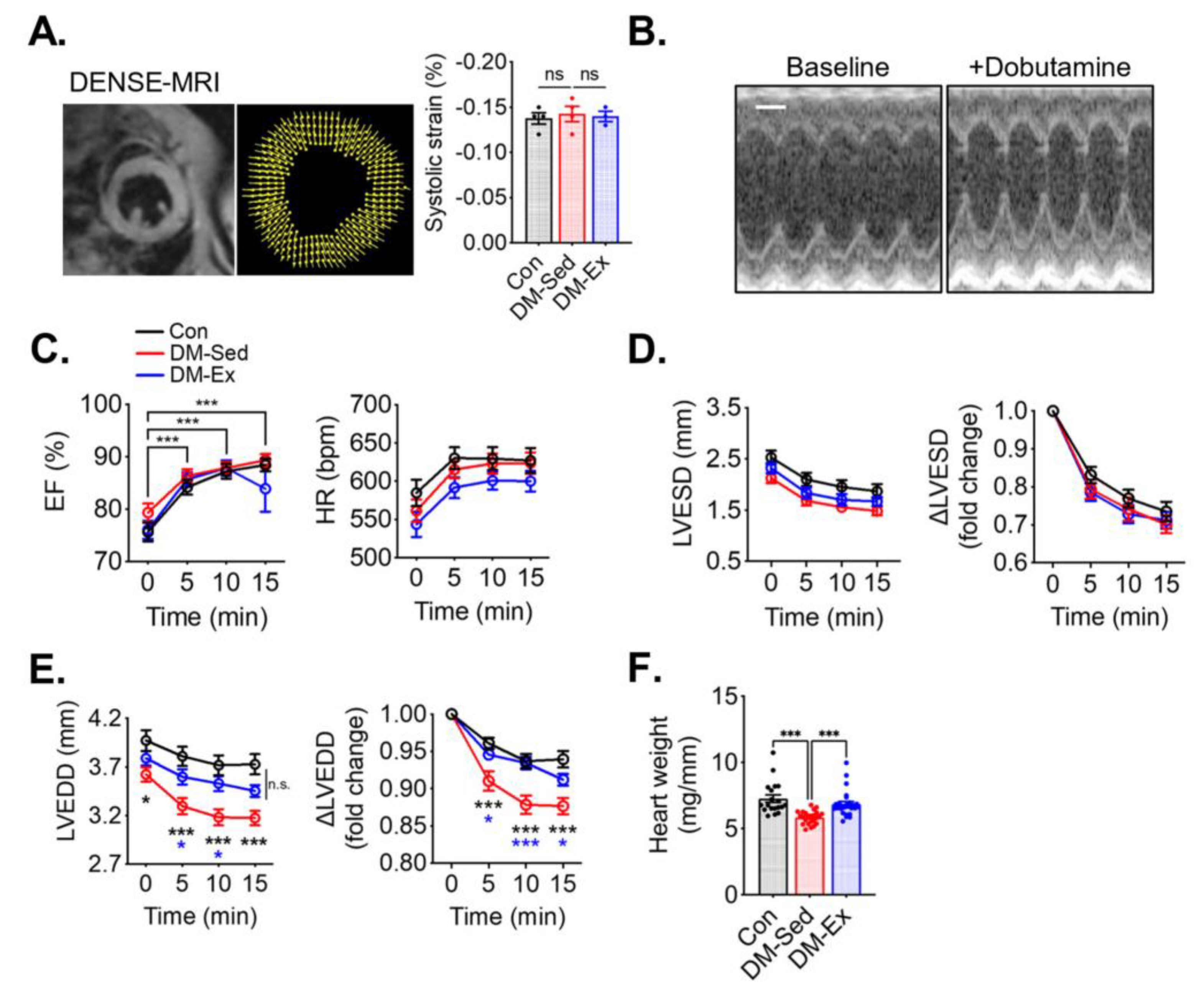
2.3. Diabetic Mice Have Preserved Systolic Function but Impaired Diastolic Function That Are Alleviated by Voluntary Wheel Running
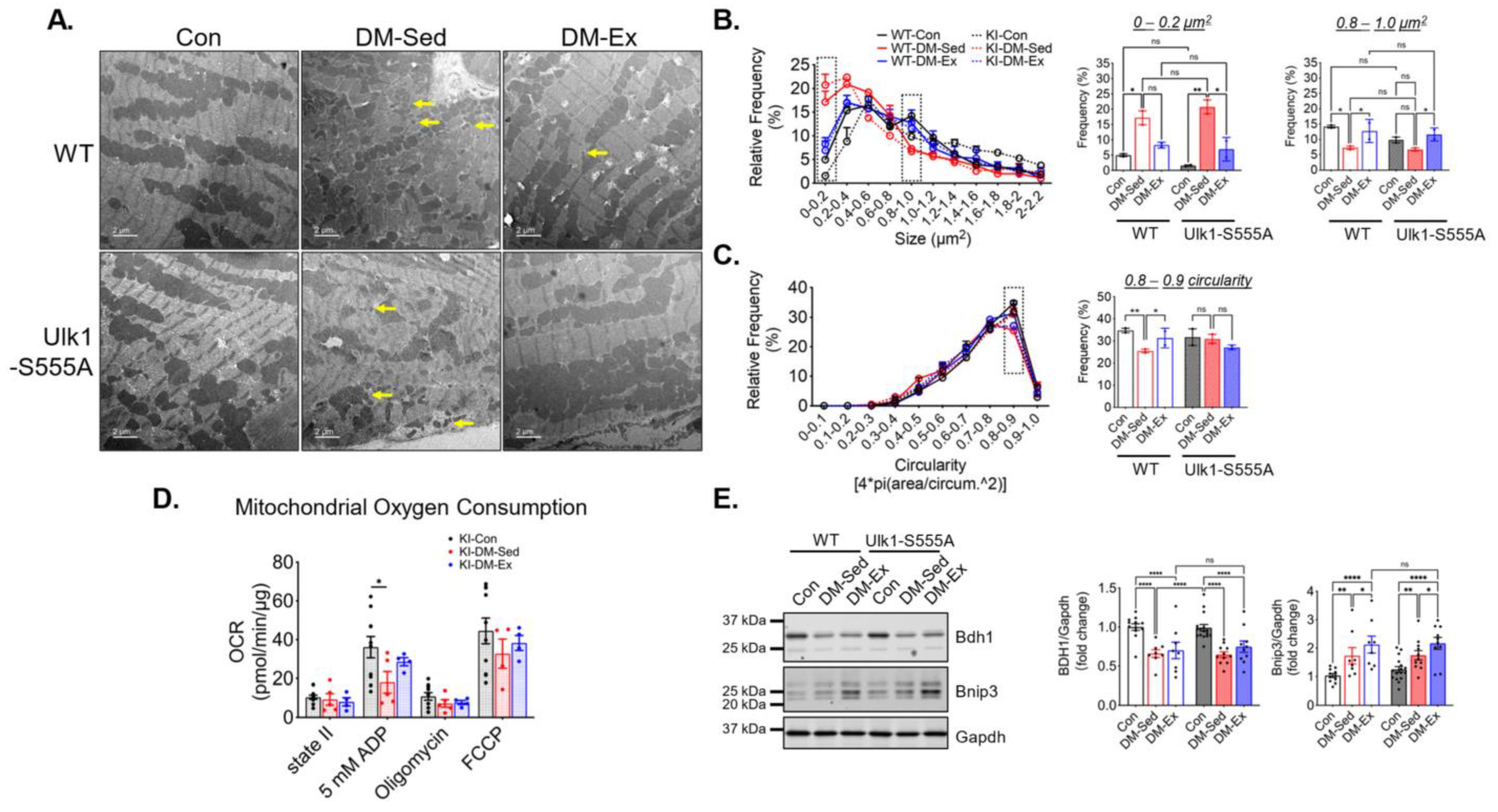
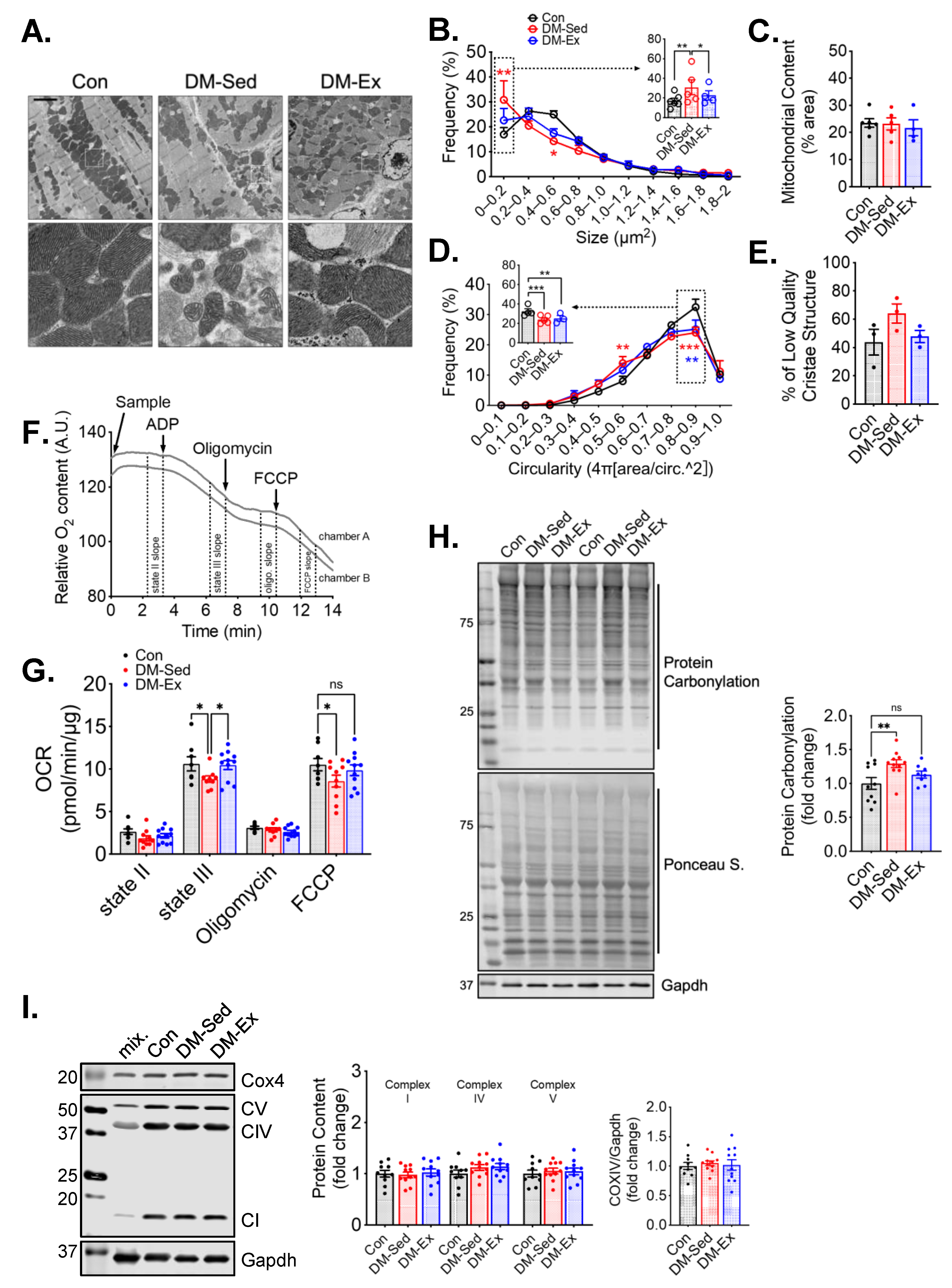
2.4. Phosphorylation of Ulk1 at S555A Is Required for Exercise-Induced Mitophagy in the Heart
2.5. Ulk1-S555A Knock-In Mice Retain the Benefits of Exercise Training in Improving Exercise Capacity under the Condition of Severe Diabetes
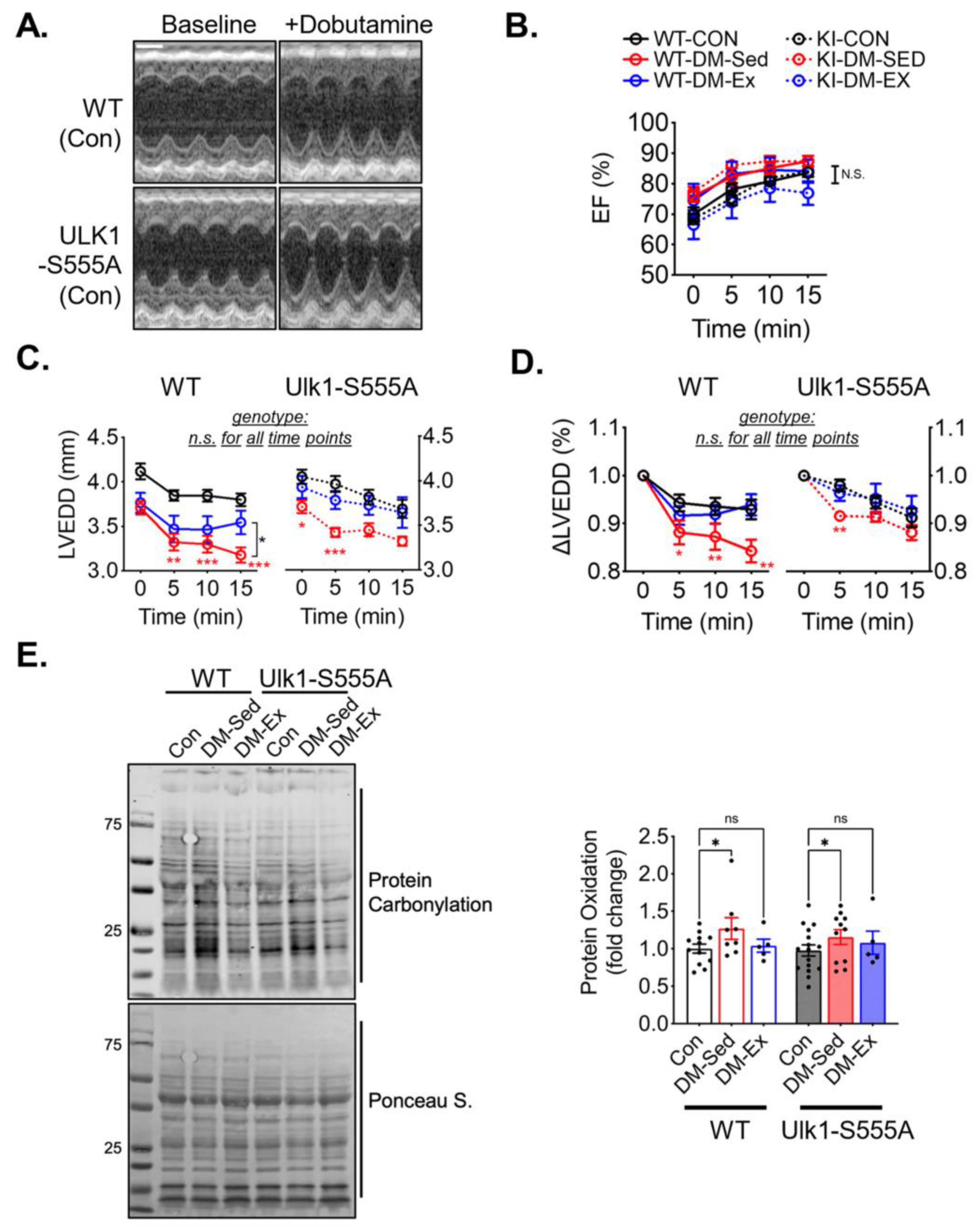
2.6. Ulk1-S555A Mice Retain Exercise-Mediated Protection against Diastolic Dysfunction under the Condition of Severe Diabetes
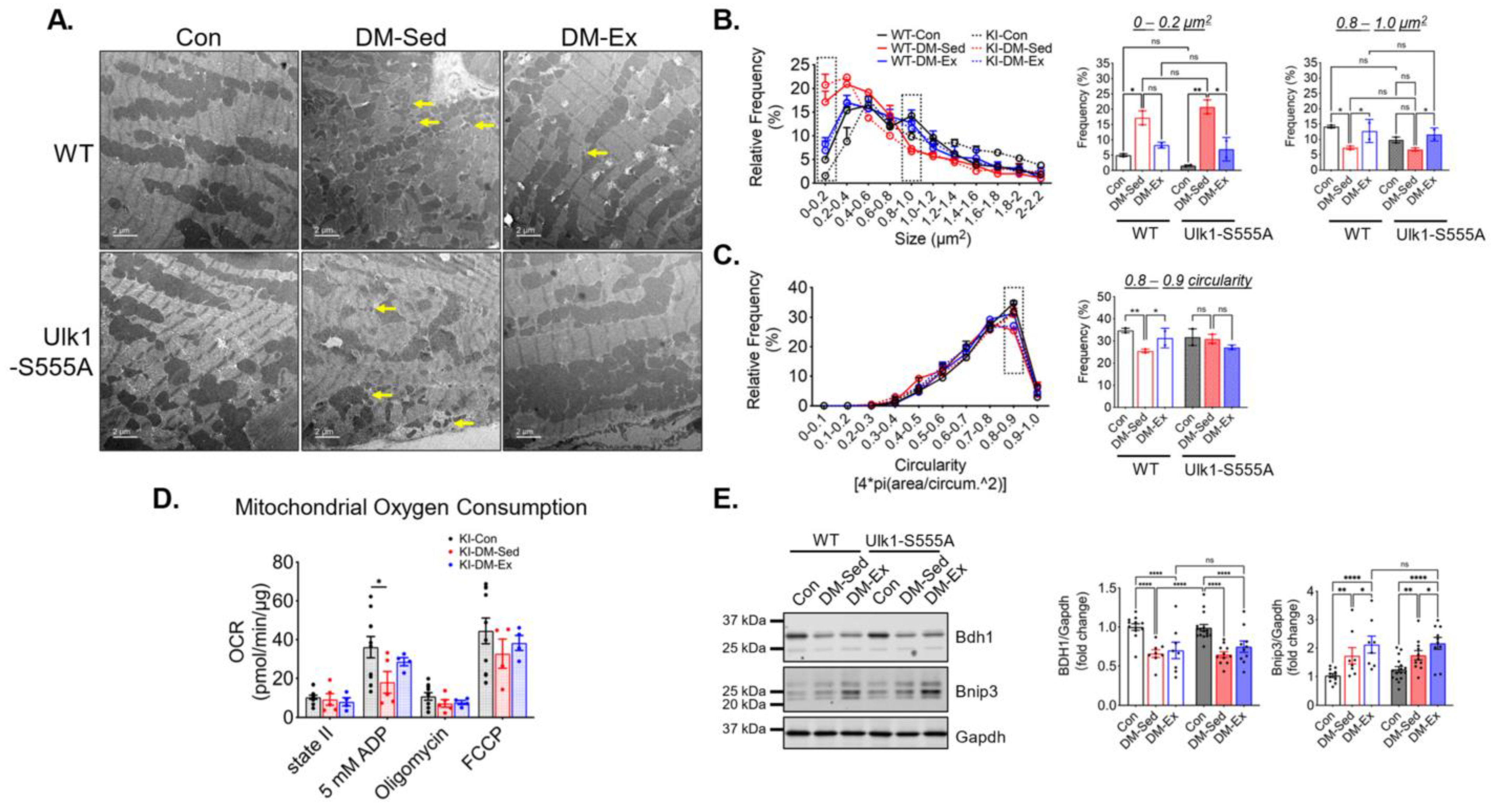
2.7. Ulk1-S555A Mice Retain Similar Mitochondrial Adaptations of Exercise in Severe Diabetes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Glucose Tolerance Test (GTT)
4.3. Dobutamine-Stress Echocardiography
4.4. Treadmill Running Test
4.5. Cardiac DENSE-MRI Analysis
4.6. Tissue Preparation
4.7. Western Blotting
4.8. Mitochondrial Oxygen Consumption Rate (OCR) Assay
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Diabetes—Overview. Available online: https://www.who.int/health-topics/diabetes#tab=tab_1 (accessed on 7 April 2023).
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Thrainsdottir, I.S.; Aspelund, T.; Thorgeirsson, G.; Gudnason, V.; Hardarson, T.; Malmberg, K.; Sigurdsson, G.; Rydén, L. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care 2005, 28, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, T.; Komorita, Y.; Peters, S.A.E.; Woodward, M. Diabetes as a risk factor for heart failure in women and men: A systematic review and meta-analysis of 47 cohorts including 12 million individuals. Diabetologia 2019, 62, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Baena-Díez, J.M.; Peñafiel, J.; Subirana, I.; Ramos, R.; Elosua, R.; Marín-Ibañez, A.; Guembe, M.J.; Rigo, F.; Tormo-Díaz, M.J.; Moreno-Iribas, C.; et al. Risk of Cause-Specific Death in Individuals With Diabetes: A Competing Risks Analysis. Diabetes Care 2016, 39, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Klionsky, D.J. Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 2008, 283, 32386–32393. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, C.; Liu, Y.; Zhao, M.; Wang, Q.; Li, P.; Liu, R.; Wai Wong, V.K.; Sun, X. L-type calcium ion channel-mediated activation of autophagy in vascular smooth muscle cells via thonningianin A (TA) alleviates vascular calcification in type 2 diabetes mellitus. Eur. J. Pharmacol. 2023, 959, 176084. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Huang, Q.; Lin, L.; Wang, L.; Wu, Y.; Wu, K.; Gao, R.; Liu, X.; Qi, L.; et al. Mitophagy disorder mediates cardiac deterioration induced by severe hypoglycemia in diabetic mice. Mol. Cell. Endocrinol. 2023, 575, 111994. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef]
- Guo, C.; Wu, R.Y.; Dou, J.H.; Song, S.F.; Sun, X.L.; Hu, Y.W.; Guo, F.S.; Wei, J.; Lin, L. Mitophagy-dependent cardioprotection of resistance training on heart failure. J. Appl. Physiol. 2023, 135, 1390–1401. [Google Scholar] [CrossRef]
- Hollekim-Strand, S.M.; Bjørgaas, M.R.; Albrektsen, G.; Tjønna, A.E.; Wisløff, U.; Ingul, C.B. High-intensity interval exercise effectively improves cardiac function in patients with type 2 diabetes mellitus and diastolic dysfunction: A randomized controlled trial. J. Am. Coll. Cardiol. 2014, 64, 1758–1760. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Boardman, N.; Aasum, E. How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1587–1605. [Google Scholar] [CrossRef]
- Pearson, M.J.; Mungovan, S.F.; Smart, N.A. Effect of exercise on diastolic function in heart failure patients: A systematic review and meta-analysis. Heart Fail. Rev. 2017, 22, 229–242. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Feng, J.; Zhang, R.; Chen, J.; Han, D.; Li, X.; Yang, B.; Fan, M.; Li, C.; Tian, Z.; et al. SIRT1 Activation by Resveratrol Alleviates Cardiac Dysfunction via Mitochondrial Regulation in Diabetic Cardiomyopathy Mice. Oxidative Med. Cell. Longev. 2017, 2017, 4602715. [Google Scholar] [CrossRef] [PubMed]
- Veeranki, S.; Givvimani, S.; Kundu, S.; Metreveli, N.; Pushpakumar, S.; Tyagi, S.C. Moderate intensity exercise prevents diabetic cardiomyopathy associated contractile dysfunction through restoration of mitochondrial function and connexin 43 levels in db/db mice. J. Mol. Cell. Cardiol. 2016, 92, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, A.; Tsukada, M.; Wada, Y.; Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997, 192, 245–250. [Google Scholar] [CrossRef]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef]
- Nah, J.; Shirakabe, A.; Mukai, R.; Zhai, P.; Sung, E.A.; Ivessa, A.; Mizushima, W.; Nakada, Y.; Saito, T.; Hu, C.; et al. Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart. Cardiovasc. Res. 2022, 118, 2638–2651. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Alternative Mitophagy Protects the Heart Against Obesity-Associated Cardiomyopathy. Circ. Res. 2021, 129, 1105–1121. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Laker, R.C.; Xu, P.; Ryall, K.A.; Sujkowski, A.; Kenwood, B.M.; Chain, K.H.; Zhang, M.; Royal, M.A.; Hoehn, K.L.; Driscoll, M.; et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014, 289, 12005–12015. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, Q.; Zhang, L.; Qiao, X.; Zhang, Y.; Tang, F.; Li, Z. Effect of CAPE-pNO2 against type 2 diabetes mellitus via the AMPK/GLUT4/GSK3β/PPARα pathway in HFD/STZ-induced diabetic mice. Eur. J. Pharmacol. 2019, 853, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lertpatipanpong, P.; Lee, J.; Kim, I.; Eling, T.; Oh, S.Y.; Seong, J.K.; Baek, S.J. The anti-diabetic effects of NAG-1/GDF15 on HFD/STZ-induced mice. Sci. Rep. 2021, 11, 15027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Deng, J.; Liu, D.; Tuo, X.; Xiao, L.; Lai, B.; Yao, Q.; Liu, J.; Yang, H.; Wang, N. Nuciferine ameliorates hepatic steatosis in high-fat diet/streptozocin-induced diabetic mice through a PPARα/PPARγ coactivator-1α pathway. Br. J. Pharmacol. 2018, 175, 4218–4228. [Google Scholar] [CrossRef] [PubMed]
- Sauvé, M.; Hui, S.K.; Dinh, D.D.; Foltz, W.D.; Momen, A.; Nedospasov, S.A.; Offermanns, S.; Husain, M.; Kroetsch, J.T.; Lidington, D.; et al. Tumor Necrosis Factor/Sphingosine-1-Phosphate Signaling Augments Resistance Artery Myogenic Tone in Diabetes. Diabetes 2016, 65, 1916–1928. [Google Scholar] [CrossRef]
- Cui, D.; Drake, J.C.; Wilson, R.J.; Shute, R.J.; Lewellen, B.; Zhang, M.; Zhao, H.; Sabik, O.L.; Onengut, S.; Berr, S.S.; et al. A novel voluntary weightlifting model in mice promotes muscle adaptation and insulin sensitivity with simultaneous enhancement of autophagy and mTOR pathway. FASEB J. 2020, 34, 7330–7344. [Google Scholar] [CrossRef]
- Li, E.; Li, X.; Huang, J.; Xu, C.; Liang, Q.; Ren, K.; Bai, A.; Lu, C.; Qian, R.; Sun, N. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 2020, 11, 661–679. [Google Scholar] [CrossRef]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef]
- O’Connor, E.; Green, S.; Kiely, C.; O’Shea, D.; Egaña, M. Differential effects of age and type 2 diabetes on dynamic vs. peak response of pulmonary oxygen uptake during exercise. J. Appl. Physiol. 2015, 118, 1031–1039. [Google Scholar] [CrossRef]
- Regensteiner, J.G.; Sippel, J.; McFarling, E.T.; Wolfel, E.E.; Hiatt, W.R. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med. Sci. Sports Exerc. 1995, 27, 875–881. [Google Scholar] [CrossRef]
- Esposito, F.; Mathieu-Costello, O.; Shabetai, R.; Wagner, P.D.; Richardson, R.S. Limited maximal exercise capacity in patients with chronic heart failure: Partitioning the contributors. J. Am. Coll. Cardiol. 2010, 55, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, B.; Taffet, G.E.; Cheng, C.P.; Kitzman, D.W. Heart failure with preserved ejection fraction in the elderly: Scope of the problem. J. Mol. Cell. Cardiol. 2015, 83, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Zhang, S.; Florido, R.; Hamo, C.; Pankow, J.S.; Michos, E.D.; Goldberg, R.B.; Nambi, V.; Gerstenblith, G.; Post, W.S.; et al. Duration of Diabetes and Incident Heart Failure: The ARIC (Atherosclerosis Risk In Communities) Study. JACC Heart Fail. 2021, 9, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Harmancey, R.; Taegtmeyer, H. The complexities of diabetic cardiomyopathy: Lessons from patients and animal models. Curr. Diab. Rep. 2008, 8, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, N.; He, H.; Chai, J.; Cheng, X.; Zhao, H.; Zhou, D.; Teng, T.; Kong, X.; Yang, Q.; et al. The zinc transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury by regulating mitophagy. Basic Res. Cardiol. 2021, 116, 54. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef]
- Shao, C.H.; Capek, H.L.; Patel, K.P.; Wang, M.; Tang, K.; DeSouza, C.; Nagai, R.; Mayhan, W.; Periasamy, M.; Bidasee, K.R. Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 2011, 60, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Stypmann, J.; Janssen, P.M.; Prestle, J.; Engelen, M.A.; Kögler, H.; Lüllmann-Rauch, R.; Eckardt, L.; von Figura, K.; Landgrebe, J.; Mleczko, A.; et al. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res. Cardiol. 2006, 101, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Ligeti, L.; Szenczi, O.; Prestia, C.M.; Szabó, C.; Horváth, K.; Marcsek, Z.L.; van Stiphout, R.G.; van Riel, N.A.; Op den Buijs, J.; Van der Vusse, G.J.; et al. Altered calcium handling is an early sign of streptozotocin-induced diabetic cardiomyopathy. Int. J. Mol. Med. 2006, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Namekata, I.; Hamaguchi, S.; Wakasugi, Y.; Ohhara, M.; Hirota, Y.; Tanaka, H. Ellagic acid and gingerol, activators of the sarco-endoplasmic reticulum Ca2+-ATPase, ameliorate diabetes mellitus-induced diastolic dysfunction in isolated murine ventricular myocardia. Eur. J. Pharmacol. 2013, 706, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bhupathy, P.; Babu, G.J.; Periasamy, M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell. Cardiol. 2007, 42, 903–911. [Google Scholar] [CrossRef]
- Quan, C.; Du, Q.; Li, M.; Wang, R.; Ouyang, Q.; Su, S.; Zhu, S.; Chen, Q.; Sheng, Y.; Chen, L.; et al. A PKB-SPEG signaling nexus links insulin resistance with diabetic cardiomyopathy by regulating calcium homeostasis. Nat. Commun. 2020, 11, 2186. [Google Scholar] [CrossRef]
- Morissette, M.P.; Susser, S.E.; Stammers, A.N.; Moffatt, T.L.; Wigle, J.T.; Wigle, T.J.; Netticadan, T.; Premecz, S.; Jassal, D.S.; O’Hara, K.A.; et al. Exercise-induced increases in the expression and activity of cardiac sarcoplasmic reticulum calcium ATPase 2 is attenuated in AMPKα. Can. J. Physiol. Pharmacol. 2019, 97, 786–795. [Google Scholar] [CrossRef]
- Wisløff, U.; Loennechen, J.P.; Falck, G.; Beisvag, V.; Currie, S.; Smith, G.; Ellingsen, O. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc. Res. 2001, 50, 495–508. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Mishra, P.K. Why the diabetic heart is energy inefficient: A ketogenesis and ketolysis perspective. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H751–H755. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079. [Google Scholar] [CrossRef] [PubMed]
- Brahma, M.K.; Ha, C.M.; Pepin, M.E.; Mia, S.; Sun, Z.; Chatham, J.C.; Habegger, K.M.; Abel, E.D.; Paterson, A.J.; Young, M.E.; et al. Increased Glucose Availability Attenuates Myocardial Ketone Body Utilization. J. Am. Heart Assoc. 2020, 9, e013039. [Google Scholar] [CrossRef] [PubMed]
- Grinblat, L.; Pacheco Bolaños, L.F.; Stoppani, A.O. Decreased rate of ketone-body oxidation and decreased activity of D-3-hydroxybutyrate dehydrogenase and succinyl-CoA:3-oxo-acid CoA-transferase in heart mitochondria of diabetic rats. Biochem. J. 1986, 240, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Altıntaş, A.; Lillard, T.S.; Zhang, M.; Connelly, J.J.; Sabik, O.L.; Onengut, S.; Rich, S.S.; Farber, C.R.; Barrès, R.; et al. Exercise during pregnancy mitigates negative effects of parental obesity on metabolic function in adult mouse offspring. J. Appl. Physiol. 2021, 130, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Dalton, N.; Mao, L.; Rockman, H.A.; Peterson, K.L.; Gottshall, K.R.; Hunter, J.J.; Chien, K.R.; Ross, J. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation 1996, 94, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Vandsburger, M.H.; French, B.A.; Kramer, C.M.; Zhong, X.; Epstein, F.H. Displacement-encoded and manganese-enhanced cardiac MRI reveal that nNOS, not eNOS, plays a dominant role in modulating contraction and calcium influx in the mammalian heart. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H412–H419. [Google Scholar] [CrossRef]
- Spottiswoode, B.S.; Zhong, X.; Hess, A.T.; Kramer, C.M.; Meintjes, E.M.; Mayosi, B.M.; Epstein, F.H. Tracking myocardial motion from cine DENSE images using spatiotemporal phase unwrapping and temporal fitting. IEEE Trans. Med. Imaging 2007, 26, 15–30. [Google Scholar] [CrossRef]
- Shah, S.A.; Reagan, C.E.; Bresticker, J.E.; Wolpe, A.G.; Good, M.E.; Macal, E.H.; Billcheck, H.O.; Bradley, L.A.; French, B.A.; Isakson, B.E.; et al. Obesity-Induced Coronary Microvascular Disease Is Prevented by iNOS Deletion and Reversed by iNOS Inhibition. Basic Transl. Sci. 2023, 8, 501–514. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, Y.; Zhang, M.; Lacy, C.; Shah, S.; Epstein, F.H.; Yan, Z. Endurance Exercise Training Mitigates Diastolic Dysfunction in Diabetic Mice Independent of Phosphorylation of Ulk1 at S555. Int. J. Mol. Sci. 2024, 25, 633. https://doi.org/10.3390/ijms25010633
Guan Y, Zhang M, Lacy C, Shah S, Epstein FH, Yan Z. Endurance Exercise Training Mitigates Diastolic Dysfunction in Diabetic Mice Independent of Phosphorylation of Ulk1 at S555. International Journal of Molecular Sciences. 2024; 25(1):633. https://doi.org/10.3390/ijms25010633
Chicago/Turabian StyleGuan, Yuntian, Mei Zhang, Christie Lacy, Soham Shah, Frederick H. Epstein, and Zhen Yan. 2024. "Endurance Exercise Training Mitigates Diastolic Dysfunction in Diabetic Mice Independent of Phosphorylation of Ulk1 at S555" International Journal of Molecular Sciences 25, no. 1: 633. https://doi.org/10.3390/ijms25010633
APA StyleGuan, Y., Zhang, M., Lacy, C., Shah, S., Epstein, F. H., & Yan, Z. (2024). Endurance Exercise Training Mitigates Diastolic Dysfunction in Diabetic Mice Independent of Phosphorylation of Ulk1 at S555. International Journal of Molecular Sciences, 25(1), 633. https://doi.org/10.3390/ijms25010633