
Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review

 ,
,  , and
, and 
Abstract
1. Introduction
2. Methods
2.1. Type of Study
2.2. Review Question
2.3. Protocol and Registration
2.4. Eligibility Criteria
2.5. Sources of Information and Search Strategy
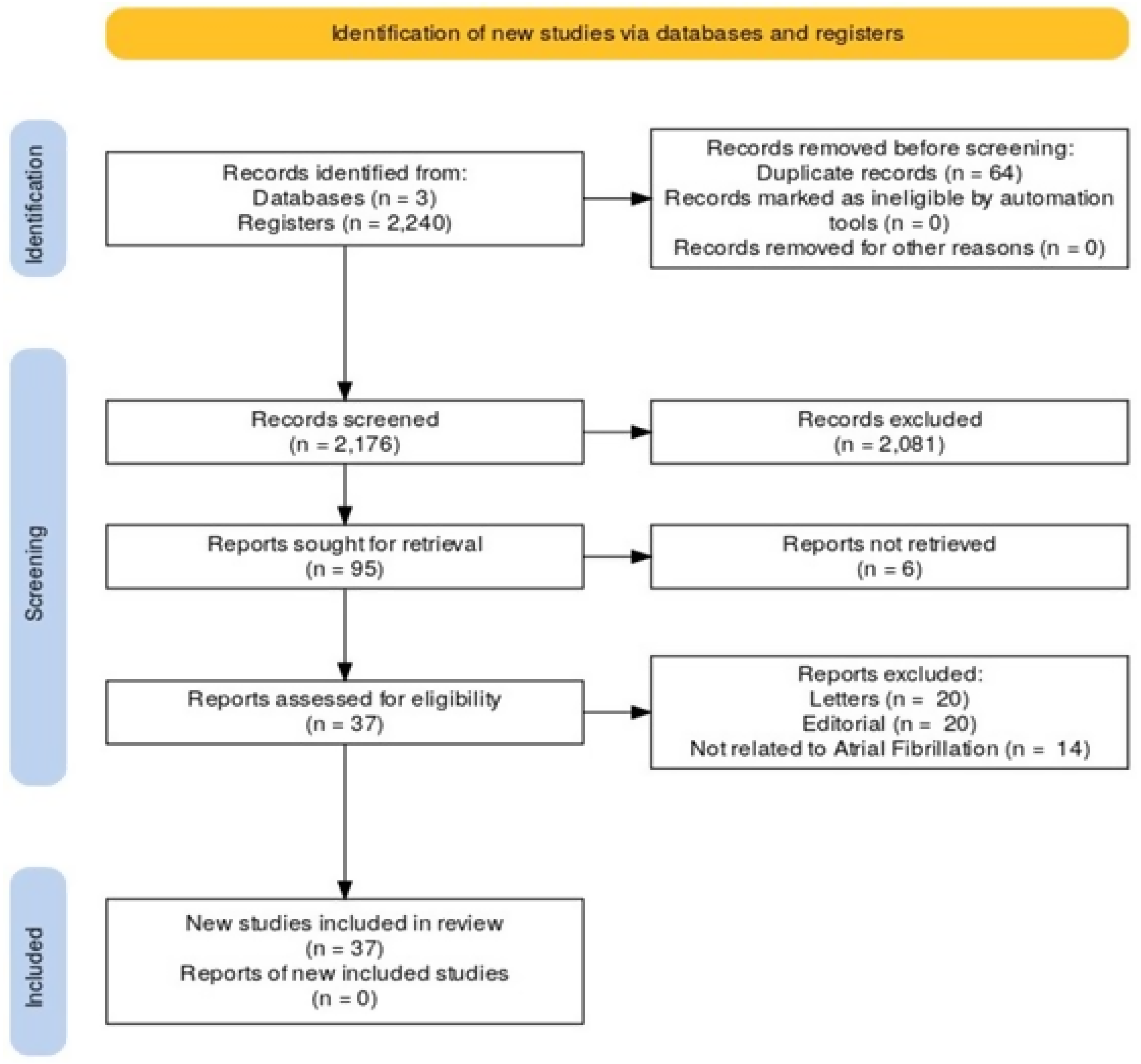
2.6. Process of Study Selection
2.7. Process of Data Extraction from Selected Studies
2.8. Risk of Bias Assessment or Quality Assessment
2.9. Data Synthesis
3. Results
3.1. Oxidative Stress
3.1.1. Mitochondrial Dysfunction
3.1.2. Electrical and Arrhythmogenic Array
3.1.3. Structural Rearrangement and Myocardial Fibrosis
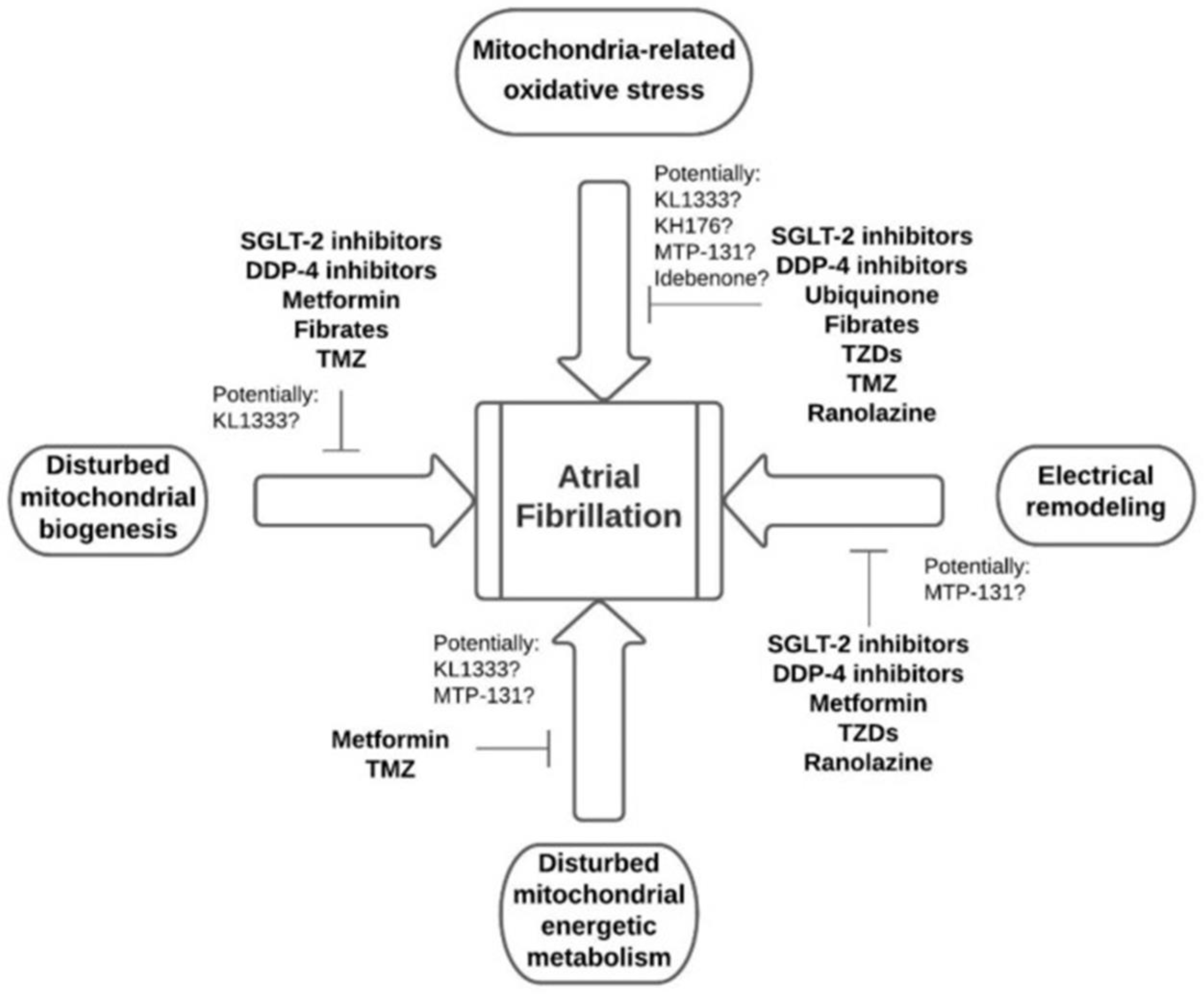
3.2. OS Modulators
3.2.1. Inflammation
3.2.2. Genetics
3.2.3. Damage to Mitochondrial DNA
3.2.4. Aging and Comorbidities
3.3. NF-KB
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- John, R.M.; Michaud, G.F.; Stevenson, W.G. Atrial fibrillation hospitalization, mortality, and therapy. Eur. Heart J. 2018, 39, 3958–3960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Meng, L.B.; Hao, M.L.; Li, X.Y.; Zou, T. CXCR4 and TYROBP mediate the development of atrial fibrillation via inflammation. J. Cell. Mol. Med. 2022, 26, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Baman, J.R.; Passman, R.S. Atrial fibrillation. JAMA 2021, 325, 2218. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Caprio, A.; Doshi, T.; Jang, S.J.; Liu, C.F.; Mosadegh, B.; Dunham, S. Multilayer fabrication of durable catheter-deployable soft robotic sensor arrays for efficient left atrial mapping. Sci. Adv. 2020, 6, eabc6800. [Google Scholar] [CrossRef] [PubMed]
- Su, K.N.; Ma, Y.; Cacheux, M.; Ilkan, Z.; Raad, N.; Muller, G.K.; Wu, X.; Guerrera, N.; Thorn, S.L.; Sinusas, A.J.; et al. Atrial AMP-activated protein kinase is critical for preventing dysregulation of electrical excitability and atrial fibrillation. JCI Insight 2022, 7, e141213. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Akazawa, H.; Tamagawa, M.; Ito, K.; Yasuda, N.; Kudo, Y.; Yamamoto, R.; Ozasa, Y.; Fujimoto, M.; Wang, P.; et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 2010, 120, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Moukabary, T.; Gonzalez, M.D. Management of atrial fibrillation. Med. Clin. N. Am. 2015, 99, 781–794. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Kolwicz Jr, S.C.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Opacic, D.; van Bragt, K.A.; Nasrallah, H.M.; Schotten, U.; Verheule, S. Atrial metabolism and tissue perfusion as determinants of electrical and structural remodelling in atrial fibrillation. Cardiovasc. Res. 2016, 109, 527–541. [Google Scholar] [CrossRef]
- Chen, J.Q.; Guo, Y.S.; Chen, Q.; Cheng, X.L.; Xiang, G.J.; Chen, M.Y.; Wu, H.L.; Huang, Q.L.; Zhu, P.L.; Zhang, J.C. TGFbeta1 and HGF regulate CTGF expression in human atrial fibroblasts and are involved in atrial remodelling in patients with rheumatic heart disease. J. Cell. Mol. Med. 2019, 23, 3032–3039. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Yu, F.; Carnes, C.A.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, Y.; Sun, A.; Peng, Y.; Hou, W.; Xiang, C.; Zhang, G.; Lai, B.; Hou, X.; Zheng, F.; et al. The coupling of mitoproteolysis and oxidative phosphorylation enables tracking of an active mitochondrial state through MitoTimer fluorescence. Redox Biol. 2022, 56, 102447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, Z.; Cao, S.; Lin, X.; Wu, M.; Li, Y.; Yin, J.; Zhou, W.; Huang, S.; Zhang, A.; et al. Reduced immunity regulator MAVS contributes to non-hypertrophic cardiac dysfunction by disturbing energy metabolism and mitochondrial homeostasis. Front. Immunol. 2022, 13, 919038. [Google Scholar] [CrossRef] [PubMed]
- Emelyanova, L.; Ashary, Z.; Cosic, M.; Negmadjanov, U.; Ross, G.; Rizvi, F.; Olet, S.; Kress, D.; Sra, J.; Tajik, A.J.; et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H54–H63. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Ascione, R.; James, A.F.; Angelini, G.D.; Suleiman, M.S. Inflammation, oxidative stress and postoperative atrial fibrillation in cardiac surgery. Pharmacol. Ther. 2015, 154, 13–20. [Google Scholar] [CrossRef]
- Zhao, Z.; Ng, C.Y.; Liu, T.; Li, H.; Li, G. Relaxin as novel strategy in the management of atrial fibrillation: Potential roles and future perspectives. Int. J. Cardiol. 2014, 171, e72–e73. [Google Scholar] [CrossRef]
- Pinho-Gomes, A.C.; Reilly, S.; Brandes, R.P.; Casadei, B. Targeting inflammation and oxidative stress in atrial fibrillation: Role of 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibition with statins. Antioxid. Redox Signal. 2014, 20, 1268–1285. [Google Scholar] [CrossRef]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Letsas, K.P.; Liu, T. Xanthine oxidase and uric acid in atrial fibrillation. Front. Physiol. 2012, 3, 150. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Hu, Q.; Chen, Y.; Zhao, L.; Zhang, J.; Bache, R.J. Acute effects of febuxostat, a nonpurine selective inhibitor of xanthine oxidase, in pacing induced heart failure. J. Cardiovasc. Pharmacol. 2006, 48, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Lin, S.Z.; Chang, N.C. Effects of urate-lowering agents on arrhythmia vulnerability in post-infarcted rat hearts. J. Pharmacol. Sci. 2016, 131, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, F.; Deng, L.; Lin, K.; Shi, X.; Zhaoliang, S.; Wang, Y. Febuxostat attenuates paroxysmal atrial fibrillation-induced regional endothelial dysfunction. Thromb. Res. 2017, 149, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, M.; Fujiki, A.; Sakamoto, T.; Nakatani, Y.; Mizumaki, K.; Inoue, H. Xanthine oxidase inhibition prevents atrial fibrillation in a canine model of atrial pacing-induced left ventricular dysfunction. J. Cardiovasc. Electrophysiol. 2012, 23, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Beneke, K.; Molina, C.E. Molecular basis of atrial fibrillation initiation and maintenance. Hearts 2021, 2, 170–187. [Google Scholar] [CrossRef]
- Muszyński, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation-Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef]
- Ren, X.; Wang, X.; Yuan, M.; Tian, C.; Li, H.; Yang, X.; Li, X.; Li, Y.; Yang, Y.; Liu, N.; et al. Mechanisms and treatments of oxidative stress in atrial fibrillation. Curr. Pharm. Des. 2018, 24, 3062–3071. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Kolettis, T.M.; Galaris, D.; Goudevenos, J.A. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol. 2007, 115, 135–143. [Google Scholar] [CrossRef]
- Schillinger, K.J.; Patel, V.V. Atrial fibrillation in the elderly: The potential contribution of reactive oxygen species. J. Geriatr. Cardiol. 2012, 9, 379–388. [Google Scholar] [CrossRef]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Urbanek, K.; De Angelis, A.; Sessa, M.; Scavone, C.; Berrino, L.; Rosano, G.M.C.; Capuano, A.; Rossi, F. Correction to: Angiotensin II and angiotensin 1–7: Which is their role in atrial fibrillation? Heart Fail. Rev. 2020, 25, 897. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Barancik, M. New aspects of pathogenesis of atrial fibrillation: Remodelling of intercalated discs. J. Physiol. Pharmacol. 2015, 66, 625–634. [Google Scholar] [PubMed]
- Pool, L.; Wijdeveld, L.F.J.M.; de Groot, N.M.S.; Brundel, B.J.J.M. The role of mitochondrial dysfunction in atrial fibrillation: Translation to druggable target and biomarker discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lim, D.S.; Lee, J.H.; Shim, W.J.; Ro, Y.M.; Park, G.H.; Becker, K.G.; Cho-Chung, Y.S.; Kim, M.K. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp. Mol. Med. 2003, 35, 336–349. [Google Scholar] [CrossRef]
- Van Wagoner, D.R. Oxidative stress and inflammation in atrial fibrillation: Role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 2008, 52, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shao, Q.; Korantzopoulos, P.; Liu, E.; Xu, G.; Li, G. Serum levels of nicotinamide-adenine dinucleotide phosphate oxidase 4 are associated with non-valvular atrial fibrillation. Biomed. Rep. 2015, 3, 864–868. [Google Scholar] [CrossRef][Green Version]
- Cangemi, R.; Celestini, A.; Calvieri, C.; Carnevale, R.; Pastori, D.; Nocella, C.; Vicario, T.; Pignatelli, P.; Violi, F. Different behaviour of NOX2 activation in patients with paroxysmal/persistent or permanent atrial fibrillation. Heart 2012, 98, 1063–1066. [Google Scholar] [CrossRef]
- Bode, D.; Semmler, L.; Oeing, C.U.; Alogna, A.; Schiattarella, G.G.; MPieske, B.; Heinzel, F.R.; Hohendanner, F. Implications of SGLT inhibition on redox signalling in atrial fibrillation. Int. J. Mol. Sci. 2021, 22, 5937. [Google Scholar] [CrossRef]
- Nishinarita, R.; Niwano, S.; Niwano, H.; Nakamura, H.; Saito, D.; Sato, T.; Matsuura, G.; Arakawa, Y.; Kobayashi, S.; Shirakawa, Y.; et al. Canagliflozin suppresses atrial remodeling in a canine atrial fibrillation model. J. Am. Heart Assoc. 2021, 10, e017483. [Google Scholar] [CrossRef]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Herrera, A.; Couselo-Seijas, M.; Feijóo-Bandín, S.; Anido-Varela, L.; Moraña-Fernández, S.; Tarazón, E.; Roselló-Lletí, E.; Portolés, M.; Martínez-Sande, J.L.; García-Seara, J.; et al. Relaxin-2 plasma levels in atrial fibrillation are linked to inflammation and oxidative stress markers. Sci. Rep. 2022, 12, 22287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, D.; Jin, Y.; Sun, G.; Lou, Q.; Wang, H.; Li, W. Costunolide ameliorates angiotensin II-induced atrial inflammation and fibrosis by regulating mitochondrial function and oxidative stress in mice: A possible therapeutic approach for atrial fibrillation. Microvasc. Res. 2023, 151, 104600. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.Z.; Murakoshi, N.; Tajiri, K.; Duo, F.; Okabe, Y.; Murakata, Y.; Yuan, Z.; Li, S.; Aonuma, K.; Song, Z.; et al. Xanthine oxidase inhibitor febuxostat reduces atrial fibrillation susceptibility by inhibition of oxidized CaMKII in Dahl salt-sensitive rats. Clin. Sci. 2021, 135, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Xu, F.; Zhu, C.; Cheng, W.K.; Li, J.; Shan, Z.L.; Li, Y.; Wang, Y.T. Effects of febuxostat on atrial remodeling in a rabbit model of atrial fibrillation induced by rapid atrial pacing Yong-Yan; et al. J. Geriatr. Cardiol. 2019, 16, 540–551. [Google Scholar] [PubMed]
- Gong, M.; Yuan, M.; Meng, L.; Zhang, Z.; Tse, G.; Zhao, Y.; Zhang, Y.; Yuan, M.; Liang, X.; Fan, G.; et al. Wenxin Keli regulates mitochondrial oxidative stress and homeostasis and improves atrial remodeling in diabetic rats. Oxid. Med. Cell. Longev. 2020, 2020, 2468031. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Cao, J.; Sun, H.; Gong, Y.; Ying, H.; Zhou, X.; Wang, Y.; Qi, C.; Yang, H.; Lv, Q.; et al. Andrographolide protects against atrial fibrillation by alleviating oxidative stress injury and promoting impaired mitochondrial bioenergetics. J. Zhejiang Univ. Sci. B 2023, 24, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Schild, L.; Keilhoff, G.; Hirte, D.; Neumann, M.; Gardemann, A.; Neumann, K.H.; Röhl, F.W.; Huth, C.; Goette, A.; et al. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp. Biol. Med. 2008, 233, 558–574. [Google Scholar] [CrossRef]
- Hadi, H.A.; Alsheikh-Ali, A.A.; Mahmeed, W.A.; Suwaidi, J.M. An Inflammatory cytokines and atrial fibrillation: Current and prospective views. J. Inflamm. Res. 2010, 3, 75–97. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, R.; Wang, X.; Li, J.; Yuan, M.; Liu, E.; Liu, T.; Li, G. Attenuation of atrial remodeling by aliskiren via affecting oxidative stress, inflammation and PI3K/Akt signaling pathway. Cardiovasc. Drugs Ther. 2021, 35, 587–598. [Google Scholar] [CrossRef]
- Xue, X.; Ling, X.; Xi, W.; Wang, P.; Sun, J.; Yang, Q.; Xiao, J. Exogenous hydrogen sulfide reduces atrial remodeling and atrial fibrillation induced by diabetes mellitus via activation of the PI3K/Akt/eNOS pathway. Mol. Med. Rep. 2020, 22, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Solus, J.; Chen, Q.; Rho, Y.H.; Milne, G.; Stein, C.M.; Darbar, D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010, 7, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Bacova, B.S.; Sykora, M.; Hlivak, P.; Barancik, M.; Tribulova, N. Mechanisms underlying antiarrhythmic properties of cardioprotective agents impacting inflammation and oxidative stress. Int. J. Mol. Sci. 2022, 23, 1416. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Fu, S.; Wei, N.; Xie, B.; Li, W.; Yang, S.; Li, Y.; Liang, Z.; Huo, H. Nitric oxide overproduction derived from inducible nitric oxide synthase increases cardiomyocyte apoptosis in human atrial fibrillation. Int. J. Cardiol. 2008, 130, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Röcken, C.; Erxleben, M.; Röhl, F.W.; Hammwöhner, M.; Huth, C.; Ebert, M.P.A.; Lendeckel, U.; Goette, A. Atrial expression of endothelial nitric oxide synthase in patients with and without atrial fibrillation. Cardiovasc. Pathol. 2010, 19, e51–e60. [Google Scholar] [CrossRef]
- Neuman, R.B.; Bloom, H.L.; Shukrullah, I.; Darrow, L.A.; Kleinbaum, D.; Jones, D.P.; Dudley, S.C. Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 2007, 53, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Kusaka, H.; Li, P.; Jackson, E.K. Effects of adenosine deaminase inhibition on blood pressure in old spontaneously hypertensive rats. Clin Exp Hypertens. 1998, 20, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Avula, U.M.R.; Dridi, H.; Chen, B.X.; Yuan, Q.; Katchman, A.N.; Reiken, S.R.; Desai, A.D.; Parsons, S.; Baksh, H.; Ma, E.; et al. Attenuating persistent sodium current–induced atrial myopathy and fibrillation by preventing mitochondrial oxidative stress. JCI Insight 2021, 6, e147371. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Tang, R.; Jiang, X.; Wang, Y.; Gu, T. Effect of TFAM on ATP content in tachypacing primary cultured cardiomyocytes and atrial fibrillation patients. Mol. Med. Rep. 2020, 22, 5105–5112. [Google Scholar] [CrossRef]
- Istratoaie, S.; Boroş, B.; Vesa, Ş.C.; Maria Pop, R.; Cismaru, G.; Pop, D.; Vasile Milaciu, M.; Ciumărnean, L.; Văcăraş, V.; Dana Buzoianu, A. Paraoxonase 1 and atrial fibrillation: Is there a relationship? Medicine 2022, 101, e31553. [Google Scholar] [CrossRef]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Han, X.; Zhang, Z.; Tse, G.; Shao, Q.; Liu, T. Role of heat shock proteins in atrial fibrillation: From molecular mechanisms to diagnostic and therapeutic opportunities. Cells 2022, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Tascanov, M.B.; Tanriverdi, Z.; Gungoren, F.; Besli, F.; Erkus, M.E.; Altiparmak, İ.H.; Gonel, A.; Koyuncu, I.; Demirbag, R.; Begenc, A.F.; et al. Relationships between paroxysmal atrial fibrillation, total oxidant status, and DNA damage. Rev. Port. Cardiol. (Engl. Ed.) 2021, 40, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Shahid, F.; Lip, G.Y.H.; Shantsila, E. Renin-angiotensin blockade in atrial fibrillation: Where are we now? J. Hum. Hypertens. 2017, 31, 425–426. [Google Scholar] [CrossRef]
- Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 9, 1292. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; de la Cruz-Ares, S.; Torres-Peña, J.D.; Alcalá-Diaz, J.F.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 and cardiovascular diseases. Antioxidants 2021, 10, 906. [Google Scholar] [CrossRef]
- Seo, K.-S.; Kim, J.H.; Min, K.N.; Moon, J.A.; Roh, T.C.; Lee, M.J.; Lee, K.W.; Min, J.E.; Lee, Y.M. KL1333, a novel NAD+ modulator, improves energy metabolism and mitochondrial dysfunction in MELAS fibroblasts. Front. Neurol. 2018, 9, 552. [Google Scholar] [CrossRef]
- Beyrath, J.; Pellegrini, M.; Renkema, H.; Houben, L.; Pecheritsyna, S.; van Zandvoort, P.; van den Broek, P.; Bekel, A.; Eftekhari, P.; Smeitink, J.A.M. KH176 safeguards mitochondrial diseased cells from redox stress-induced cell death by interacting with the thioredoxin system/peroxiredoxin enzyme machinery. Sci. Rep. 2018, 8, 6577. [Google Scholar] [CrossRef]
- Gao, G.; Dudley, S.C., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid. Redox Signal. 2009, 11, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Pauklin, P.; Zilmer, M.; Eha, J.; Tootsi, K.; Kals, M.; Kampus, P. Markers of inflammation, oxidative stress, and fibrosis in patients with atrial fibrillation. Oxid. Med. Cell. Longev. 2022, 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.S.H.; Jerkins, T. In praise of pioglitazone: An economically efficacious therapy for Type 2 diabetes and other manifestations of the metabolic syndrome. Diabetes Obes. Metab. August 2023, 25, 3093–3102. [Google Scholar] [CrossRef]
- Godoy-Marín, H.; Duroux, R.; Jacobson, K.A.; Soler, C.; Colino-Lage, H.; Jiménez-Sábado, V.; Montiel, J.; Hove-Madsen, L.; Ciruela, F. Adenosine A2a receptors are upregulated in peripheral blood mononuclear cells from atrial fibrillation patients. Int. J. Mol. Sci. 2021, 22, 3467. [Google Scholar] [CrossRef]
- Lin, P.H.; Lee, S.H.; Su, C.P.; Wei, Y.H. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic. Biol. Med. 2003, 35, 1310–1318. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author/ Year | Country | Title | Type of Study | Study Population | Objectives | Methods | Main Findings |
|---|---|---|---|---|---|---|---|
| Emelyanova 2016 [15] | USA | Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation | Case-control | Right atrial tissue between non-AF and AF patients undergoing open-heart surgery. | The study compared mitochondrial oxidative phosphorylation (OXPHOS) complexes and oxidative stress. | Between 2012 and 2016, elective open-heart surgery patients were examined for atrial appendage tissues, examining factors like O2 oxidoreductase activity, mitochondrial OXPHOS complexes, citrate synthase activity, and protein expression. | AF is linked to decreased ETC activity and increased oxidative stress, which can contribute to the progression of the substrate for AF. |
| Muszyński. 2021 [27] | Switzerland | Mitochondrial dysfunction in atrial fibrillation—mechanisms and pharmacological interventions | Review | Atrial fibrillation | The study seeks to review mitochondrial dysfunction mechanisms in atrial fibrillation, exploring their role and potential therapeutic implications, aiming to enhance our understanding and contribute to improved prevention and treatment strategies for arrhythmia. | This text aims to comprehend the pathophysiological mechanism of arrhythmia, its prevention methods, and the role of mitochondrial dysfunction in the onset of arrhythmia. | Mitochondrial dysfunction, a key factor in cardiomyocyte integrity and cardiac performance, can be addressed through medications that target energetic imbalance, metabolic disturbance, and oxidative stress. |
| Ren, 2018 [28] | China | Mechanisms and treatments of oxidative stress in atrial fibrillation | Review | Atrial fibrillation | The study aims to review pathways of reactive oxygen species (ROS) induction in atrial fibrillation (AF), focusing on electrical and structural remodeling. It explores sources and factors contributing to AF-related oxidative stress, identifying potential therapeutic targets through scavenging oxidative stress markers. | The research uses atrial electrical and structural remodeling data to explore various reactive oxygen species pathways that can induce atrial fibrillation (AF). | AF-related oxidative stress, triggered by factors like NADPH oxidase activation, calcium overload, and cardiovascular conditions, may be a therapeutic target for AF. |
| Korantzo poulos. 2007 [29] | Greece | The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation | Review Article | Atrial fibrillation | This summary critically analyzes the current literature on AF and oxidative stress, highlighting the scientific background of antioxidant therapeutic interventions. | The authors conducted a comprehensive search using the MEDLINE database, reference lists, and significant cardiovascular journal supplements to identify studies published up to December 2005. | Oxidative stress is linked to AF pathophysiology, contributing to arrhythmia perpetuation. Modulating oxidative stress can benefit AFs development of inflammatory and electrophysiological changes. |
| Schillinger, 2012 [30] | USA | Atrial fibrillation in the elderly: The potential contribution of reactive oxygen species | Review | Atrial fibrillation | The study explores the link between aging and AF through reactive oxygen species’ (ROS) oxidative damage, which induces intracellular and extracellular changes. | An overview of AF pathophysiology and introduces the critical structures that predispose an otherwise healthy atrium to AF when damaged. The available evidence that ROS can lead to damage to these vital structures is then reviewed. Finally, the evidence linking the aging process to the pathogenesis of AF is discussed. | To be certain, through serving as agents of oxidative cellular damage, ROS is likely to be a major causative factor in the development of AF. The cellular changes brought about by ROS-mediated damage are sufficient to promote tissue changes consistent with AF triggers and atrial electrical and structural remodeling. |
| Mason, 2020 [31] | Germany | Cellular and mitochondrial mechanisms of atrial fibrillation | Review | Atrial fibrillation | This review discusses the importance of mitochondrial Ca2+ handling in regulating ATP production and mitochondrial ROS emission and how alterations may play a role in AF, particularly in these aspects of mitochondrial activity. | It involves understanding the molecular mechanisms underlying AF and specific treatment options. | In the current review, we discussed the hypothesis that remodeling and increased energy demand during AF lead to oxidative stress, shifting the redox environment to a state of energy deficit and compromised ROS scavenging capacity. |
| Mascolo, 2020 [32] | Italy | Angiotensin II and angiotensin 1–7: which is their role in atrial fibrillation? | Review | Atrial fibrillation | This review aims to provide an overview of the evidence for the possible role of the two RAS pathways (classic and non-classic) in the pathophysiology of AF, the proposed cellular and molecular mechanisms, and the results of clinical studies with classic RAS antagonists. | Understand the role of the RAS in the induction of AF, influenced by inflammatory and cardiac processes, electrical remodeling, and epicardial fat accumulation. | In this review, we summarized the evidence showing that both RAS pathways can balance the onset of AF through different biological mechanisms involving inflammation, epicardial adipose tissue (EAT) accumulation, and cardiac remodeling. |
| Tribulova, 2015 [33] | Slovak Republic | New aspects of the pathogenesis of atrial fibrillation: Remodeling of intercalated discs | Review | Atrial fibrillation | Understanding the role of key factors of aging, oxidative stress, and inflammation in the development of age-related cardiovascular disease and AF. | Review of reactive oxygen species, their production, and relationship with systemic inflammation. Approach to studies that consider cardiac structural and electrophysiological remodeling as crucial for developing and maintaining atrial fibrillation. | Data suggest that alterations in atrial connexin-43 and/or connexin-40 expression, phosphorylation, and distribution affect cell-to-cell electrical coupling and molecular signaling that is proarrhythmogenic. Gap junctional connexin channels are considered targets for arrhythmia prevention, and therapeutic interventions for mitochondria-related reactive oxygen species appear. In addition, aging is accompanied by abnormalities in adhesive junctions that most likely promote asynchronous contractions and arrhythmias. |
| Pool, 2021 [34] | Switzer land | The role of mitochondrial dysfunction in atrial fibrillation: translation to druggable targets and biomarker discovery | Review | Atrial fibrillation | To dissect molecular mechanisms that drive AF. Investigating the role of mitochondrial impairment in AF may guide the path towards new therapeutic and diagnostic targets. | A review of the molecular mechanisms that drive AF and the genesis of this clinical situation. | Novel findings show a key role for mitochondrial dysfunction in the onset and progression of AF. Current therapeutic strategies for AF are aimed directly at the suppression of AF symptoms but are not effective in terms of preventing AF progression. Furthermore, no AF-specific biomarkers are available. So far, several mitochondrial biomarkers have been tested in clinical AF. Recent findings indicate the potential diagnostic value of blood-based 8-OHdG and cfc-mtDNA in staging AF. |
| Kim, Y. H., 2003 [35] | Korea | Gene expression profiling of oxidative stress on atrial fibrillation in humans | Case-control | Human myocardial tissues under AF and oxidative stress | The study aims to examine the gene transcriptional profiles in human myocardial tissues under AF and oxidative stress conditions. | Researchers studied the effects of oxidative stress on atrial fibrillation (AF) using radioactive DNA microarrays to evaluate changes in gene expression in 26 AF patients undergoing the Maze procedure. | Gene expression profiles reveal 30 upregulated and 25 downregulated genes in AF patients, with five ROS-related genes increasing by over 2.0 and two antioxidant genes decreasing. |
| Van Wagoner, D. R, 2008 [36] | USA | Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target | Review | Atrial fibrillation | The study explores the potential of therapeutic intervention in manipulating oxidative and inflammatory pathways. | This review examines the various treatments for AF utilizing ion channel blockade and the fundamental mechanisms that underlie AF. | AF is a multifactorial arrhythmia characterized by rapid atrial contractile activity degradation, blood stasis, thrombus formation, and increased electrical instability due to aging and other risk factors. |
| Liu 2015 [37] | China | Serum levels of nicotinamide adenine dinucleotide phosphate oxidase 4 are associated with non-valvular atrial fibrillation. | Clinical study | Atrial fibrillation | The present study aimed to investigate the potential association between serum levels of NOX4 and inflammatory biomarkers and AF. | The study involved 180 consecutive AF patients admitted to Tianjin Medical University’s Department of Cardiology between 2012 and 2013, with a final population of 108 consecutive AF patients. | The study indicates a correlation between elevated NOX4 levels and AF, indicating a potential role of NOX4 in the pathophysiology of human AF. |
| Cangemi, 2009 [38] | Italy | Different behavior of NOX2 activation in patients with paroxysmal/persistent, or permanent atrial fibrillation | Case-control | Patients with atrial fibrillation and the Patient Control Group. | To define the role of NOX2 and isoprostanes, a marker of oxidative stress, in the different settings of AF. | A study involving 174 patients with AF and 90 controls was conducted, measuring urinary isoprostanes and serum levels of soluble NOX2-derived peptides. | Patients with paroxysmal/persistent AF had higher urinary isoprostane and sNOX2-dp concentrations than permanent AF and controls, with baseline values independently associated with AF type. |
| David Bode 2021 [39] | Germany | Implications of SGLT Inhibition on Redox Signaling in Atrial Fibrillation | Review | Atrial fibrillation | The study evaluates the implications of SGLT inhibition on redox signaling in AF. | This study involved a review of clinical data and trials, exploring the association between diabetes, AF, and the use of SGLTi. | The study findings indicate that SGLTi has potential benefits in reducing AF burden. |
| Ryo Nishinarita, 2021 [40] | Japan | Canagliflozin Suppresses Atrial Remodeling in a Canine Atrial Fibrillation Model | Experimental study | Beagle dogs undergoing rapid atrial pacing | The study explored the potential benefits of Canagliflozin (CAN) and other SGLT2 inhibitors in preventing atrial fibrillation (AF) and inhibiting atrial remodeling promotion. | The study involved 12 beagle dogs, 10 undergoing rapid atrial pacing, and compared their performance over three weeks, analyzing parameters and histological findings. | CAN treatment reduces atrial-effective refractory period, conduction velocity, and atrial artery disease (AF) incidence, mitigating interstitial fibrosis and oxidative stress in atrial tissues. |
| Koizumi; 2023 [41] | Japan | Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. | Experimental study | Type-2 diabetic rat’s atrium | The study investigated the potential of empagliflozin to reduce mitochondrial reactive oxygen species (ROS) generation and reduce fibrosis in diabetic patients, considering the correlation between oxidative stress and AF pathogenesis. | The study examined the effects of empagliflozin on atrial mitochondrial respiratory capacity, reactive oxygen species generation, oxidative stress markers, protein expression, atrial tachyarrhythmia inducibility, and fibrosis in a type-2 diabetes model. | The study suggests empagliflozin may have cardioprotective effects by reducing mitochondrial ROS generation in diabetic rats’ atrium, potentially suppressing the development of atrial fibrillation (AF) in type-2 diabetes. |
| Aragón- Herrera, 2022 [42] | Spain | Relaxin-2 plasma levels in atrial fibrillation are linked to inflammation and oxidative stress markers. | Case-control | Caucasian patients with persistent AF and Patient Control Group | The study investigates the correlation between relaxin-2 plasma levels in the left atrium and peripheral vein with fibrosis, inflammation, and oxidative stress in AF patients and its anti-fibrotic properties. | The study involved 68 Caucasian patients with persistent AF who underwent pulmonary vein radiofrequency catheter ablation at the University Clinical Hospital of Santiago de Compostela. | Patients with higher relaxin-2 concentrations in peripheral veins had higher Gal-3 levels in plasma, and RLX2 treatment reduced mRNA expression levels in NHCF-A cells. |
| Liu, 2023 [43] | China | Costunolide ameliorates angiotensin II-induced atrial inflammation and fibrosis by regulating mitochondrial function and oxidative stress in mice: a possible therapeutic approach for atrial fibrillation. | Experimental study | Male C57BL/6 mice induced with AF | The study examines the positive impact of costunolide on angiotensin 2-induced atrial fibrillation. | Male C57BL/6 mice induced with AF using Ang II were administered varying doses of Costunolide (COS) for three weeks. | Costunolide has shown potential therapeutic benefits in treating Angiotensin II-induced atrial fibrillation by reducing inflammation, fibrosis, and mitochondrial dysfunction. |
| Xu, 2021 [44] | Japan | The xanthine oxidase inhibitor febuxostat reduces atrial fibrillation susceptibility by inhibiting oxidized CaMKII in Dahl salt-sensitive rats. | Experimental study | Dahl salt-sensitive rats | The study evaluated the impact of febuxostat, an XO inhibitor, on salt-induced hypertension in a rat model compared to allopurinol. | Researchers studied Dahl salt-sensitive rats on high-salt diets, dividing them into three groups and administering treatments orally. They measured blood pressure, atrial fibrillation, and protein expression. | Febuxostat and allopurinol significantly reduced hypertension-related atrial fibrillation in rats, improving calcium handling. XO inhibitors reduced Ca2+ handling protein expression and partially restored connexin 40 expressions. |
| Yong-Yan Fan; 2019 [45] | China | Effects of febuxostat on atrial remodeling in a rabbit model of atrial fibrillation induced by rapid atrial pacing | Experimental study | Rabbits with different RAP levels | The study evaluated the impact of febuxostat on atrial remodeling in a rabbit model of atrial fibrillation (AF) induced by rapid atrial pacing and explored its mechanisms. | Rabbits were divided into four groups, each with different RAP levels. The effects of febuxostat on atrial remodeling, inflammation, oxidative stress markers, and left atrium signaling pathways were examined. | Rapid atrial pacing in rabbits leads to atrial enlargement, dysfunction, and fibrillation, while Febuxostat treatment suppresses these changes by inhibiting atrial electrical and structural remodeling. |
| Gong; 2020 [46] | China | Wenxin Keli Regulates Mitochondrial Oxidative Stress and Homeostasis and Improves Atrial Remodeling in Diabetic Rats | Experimental study | Atrial fibroblasts isolated from neonatal Sprague-Dawley (SD) rats | This study evaluated the hypothesis that WXKL can improve atrial remodeling in diabetic rats, restoring mitochondrial function. | Primary cultures of atrial fibroblasts isolated from neonatal Sprague-Dawley (SD) rats were used. Male SD rats were divided into control, DM (diabetes mellitus), and DM + WXKL (WXKL treatment) groups. Diabetes induction was performed by injection of STZ, followed by treatment with WXKL. | WXKL prevents oxidative stress and improves mitochondrial function. In diabetic rats treated with WXKL, several parameters were improved, including atrial fibrosis, reduced atrial diameter, increased atrial conduction velocity, and reduced induction of atrial fibrillation. |
| YU, 2023 [47] | China | Andrographolide protects against atrial fibrillation by alleviating oxidative stress injuries and promoting impaired mitochondrial bioenergetics | Experimental study | HL-1 cells and rabbits | This study aimed to explore the mechanisms of action of andrographolide on AF. | The study investigated Andr’s role in atrial fibrillation (AF) by pre-treating HL-1 cells and rabbits with Andr before RES and atrial pacing using RNA sequencing. | Andrographolide effectively mitigates rapid atrial pacing, causing changes in electrophysiology, inflammation, oxidative damage, and apoptosis, potentially through a therapeutic mechanism involving the Keap1-Nrf2 complex. |
| Bukowska, 2007 [48] | Germany | Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia | Case-control | Ex vivo atrial tissue from patients with and without atrial fibrillation | The study investigates the impact of AF on mitochondrial dysfunction and oxidative stress-activated signal transduction by analyzing NF-kB, LOX-1, ICAM-1, and HO-1. | Ex vivo atrial tissue from patients with and without atrial fibrillation was studied for mitochondrial structure and respiration, while NF-kB target gene expression was measured using various methods. | Oxidative stress, mitochondrial structure, and respiration were observed in human atrial tissue slices, with NF-jB accumulation and elevated ICAM-1 expression. A blockade with verapamil prevented these changes. |
| Hadi, 2010 [49] | United Arab Emirates | Inflammatory cytokines and atrial fibrillation: current and prospective views | Post-hoc comparison of data collected in a prospective randomized investigation | Atrial fibrillation | To present an overview of the evidence linking inflammatory cytokines to AF. | The authors analyzed articles published until December 2009 on Medline, Pubmed, Scopus, and EBSCOhost® using indexing terms for inflammation, cytokines, AF, and atrial arrhythmias. | Inflammatory cytokines and markers like IL-6 and CRP are linked to atrial fibrillation (AF), potentially indicating inflammation and predicting thromboembolic events in AF patients. |
| Zhao, 2020 [50] | China | Attenuation of atrial remodeling by aliskiren via affecting oxidative stress, inflammation, and the PI3K/Akt signaling pathway | Experimental study | Dogs subjected to rapid atrial pacing | The study investigates the cardioprotective effect of aliskiren (ALS) and its potential molecular mechanisms in atrial remodeling, focusing specifically on atrial fibrillation (AF). | The study involved acute and chronic experiments on dogs subjected to rapid atrial pacing, assessing parameters like effective refractory periods, AF inducibility, and average duration. | Aliskiren has shown cardioprotective effects by reducing electrophysiological alterations, oxidative stress, inflammation, and atrial remodeling, possibly through regulating the PI3K/Akt signaling pathway. |
| Xue, 2020 [51] | China | Exogenous hydrogen sulfide reduces atrial remodeling and atrial fibrillation induced by diabetes mellitus via activation of the PI3K/Akt/eNOS pathway | Experimental study | Sprague-Dawley rats | This study aimed to explore the impact of hydrogen sulfide on diabetes mellitus-induced atrial fibrillation and its underlying mechanisms. | The study involved Sprague-Dawley rats in four groups: control, DM, H2S, and DM + H2S, analyzing atrial fibrillation, fibrosis, protein expression, cell viability, and cardiac fibroblast migration. | H2S may reduce atrial fibrosis and DM-induced AF by activating the PI3K/Akt/eNOS pathway. |
| Li, 2010 [52] | EUA | Role of inflammation and oxidative stress in atrial fibrillation | Case-control | Patients with and without AF | The study aims to understand the role of inflammation and oxidative stress in developing AF. | A study compared 305 AF patients with and without AF, assessing serum inflammatory markers and oxidative stress and comparing them to control patients. | IL-6, IL-8, IL-10, TNF-α, MCP1, VEGF, and NTpBNP concentrations were linked to AF, with graded increases in TNF-α and NTpBNP among paroxysmal, persistent, and permanent AF subgroups. |
| Andelova, 2022 [53] | Slovakia | Mechanisms Underlying the Antiarrhythmic Properties of Cardioprotective Agents Impacting Inflammation and Oxidative Stress | Review | Atrial fibrillation | The study aimed to examine the antiarrhythmic efficacy and molecular mechanisms of current clinically used pharmaceuticals in the context of AF. | The study examines the biomolecular mechanisms of FA and the therapeutic efficacy of Sodium-Glucose Cotransporter-2 Inhibitors, Statins, and Omega-3 fatty acids in preventing oxidative and inflammatory stress. | The approach suggests that reducing oxidative and inflammatory stress can eliminate pro-arrhythmic factors and arrhythmia substrates, making it a potent tool for reducing cardiac arrhythmia burden. |
| Han, 2008 [54] | China | Nitric oxide overproduction derived from inducible nitric oxide synthase increases cardiomyocyte apoptosis in human atrial fibrillation | Case-control | patients with permanent AF and sinus rhythm after mitral valve replacement surgery | The study aims to investigate the potential role of iNOS in atrial remodeling in AF. | The study investigated patients with permanent AF and sinus rhythm after mitral valve replacement surgery, using Western blotting, immunohistochemical staining, and the NOX Detection Kit to measure cardiac function. | NOS expression in the right atrium was upregulated, while eNOS did not. Inflammation and oxidative damage in the right atrium of AF patients were associated with increased iNOS/eNOS expression. |
| Bukowska, 2010 [55] | Germany | Atrial expression of endothelial nitric oxide synthase in patients with and without atrial fibrillation | Case-control | atrial tissue from 234 patients with atrial fibrillation | The study aims to assess the endocardial expression of eNOS in atrial tissue samples from patients with and without atrial fibrillation (AF). | Tissue microarrays were used to analyze atrial tissue from 234 patients, examining differences in atrial eNOS expression, with immunohistological results confirmed by Western blotting in selected patients. | eNOS expression is influenced by factors like diabetes mellitus and coronary artery disease, with women with AF having the lowest levels. |
| Neuman, 2007 [56] | USA | Oxidative stress markers are associated with persistent atrial fibrillation | Cross-sectional study | males with or without AF | To compare serum markers of oxidation and associated inflammation in individuals with or without AF. | A cross-sectional study compared serum markers of oxidative stress and inflammation in 40 males with or without AF, matched by age, sex, diabetes, and smoking status. | Oxidative stress, not inflammatory markers, is statistically associated with AF, suggesting that oxidative stress markers may be predictive in AF management. |
| Pinho- Gomes, 2014 [57] | United Kingdom | Targeting inflammation and oxidative stress in atrial fibrillation: Role of 3-hydroxy-3-methylglutaryl-coenzyme reductase inhibition with statins | Review | Atrial fibrillation | Using statins to decrease inflammation by restoring the myocardial nitroso-redox balance. | This review explores articles discussing potential statin-related therapies and their potential to reduce inflammation. | Statins show the highest anti-arrhythmic benefits in preventing postoperative AF but limited benefits in primary AF prevention, making them unsuitable for preventing incident AF or recurrence. |
| Godoy- Marín, H.; 2021 [58] | Spain | Adenosine a2a receptors are upregulated in peripheral blood mononuclear cells from atrial fibrillation patients | Case-control | Samples from patients with sinus rhythms and atrial fibrillation | The study explores the expression of adenosine A2A receptor (A2AR) in right atrium biopsies and peripheral blood mononuclear cells from non-dilated sinus rhythm (ndSR), dilated sinus rhythm (dSR), and AF patients. | Samples from patients with sinus rhythms and atrial fibrillation were collected and analyzed using various methods, including gel electrophoresis, immunoblotting, RT-qPCR, cell culture, Flow Cytometry, and Confocal Imaging. | The study found increased A2AR expression in the right atrium of AF patients, with adenosine content and reduced ADA activity in plasma, and a positive correlation between A2AR expression and PBMCs. |
| Avula, 2021 [59] | USA | Attenuating persistent sodium current-induced atrial myopathy and fibrillation by preventing mitochondrial oxidative stress | Experimental study | crossbreeding mice expressing persistent sodium channels and mice expressing human mitochondrial catalase (mCAT) | This study aims to comprehend the mechanisms influencing structural and electrophysiological remodeling in the atria due to an increased persistent sodium current. | The study involved crossbreeding mice expressing persistent sodium channels (NaV1.5 F1759A) with mice expressing human mitochondrial catalase (mCAT). | mCAT expression reduced mitochondrial oxidative stress, atria structural changes, atrial fibrillation, and ryanodine receptor dysfunction, reducing spontaneous and stimulation-induced atrial fibrillation. |
| Lin, 2003 [60] | China | Oxidative damage to mitochondrial DNA in the atrial muscle of patients with atrial fibrillation | Case-control | patients with chronic AF | The authors utilized long-range polymerase chain reaction (PCR) to detect large-scale deletions of mtDNA in the atrial muscle of AF patients. | Right atrial appendages were removed from patients with chronic AF and sinus rhythm during heart surgery, and cellular DNA was extracted, revealing large-scale deletions. | The study found a high frequency of large-scale mtDNA deletions, particularly the 4977-bp deletion, in patients with atrial fibrillation (AF), with oxidative damage causing more significant damage. |
| Istratoaie, 2022 [61] | USA | Paraoxonase 1 and atrial fibrillation: Is there a relationship? | Case-control | patients with symptomatic paroxysmal or persistent AF and the patient control group | The study aims to assess the concentration and activity of PON1 arylesterase (AREase) in patients with AF. | The study analyzed 67 patients with symptomatic paroxysmal or persistent AF admitted for cardioversion and 59 without AF, evaluating clinical parameters, lipid profile, PON1 concentration, and AREase. | Oxidative stress contributes to diseases like arrhythmias and increased risk of atrial fibrillation (AF), promoting endocardial dysfunction, left atrial thrombus, and stroke and influencing the efficacy of various drugs. |
| Samman, 2017 [62] | USA | Association between oxidative stress and atrial fibrillation | Prospective study | coronary angiography patients | The study hypothesized that prevalent and incident AF are associated with glutathione (EhGSH) and cysteine redox potentials, estimating systemic oxidative stress. | Aminothiol plasma levels in 1439 coronary angiography patients, including 148 with AF diagnoses, showed an 11.5% incidence of AF in 104 out of 917 patients after 6.3 years. | EhGSH levels in CAD patients increase the risk of prevalent and incident AF, independent of hsCRP level and other AF predictors, and correlate with the CHA2DS2-VASc score. |
| KL1333 | Increases mitochondrial activity and reduces oxidative stress in fibroblasts in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like events. It also increases NAD+ levels and stimulates sirtuin 1/AMP-activated protein kinase/peroxisome proliferator-activated receptor-gamma coactivator 1alphasignaling [69]. |
| KH176 | By interacting with the thioredoxin system and the enzymatic mechanism of peroxiredoxin, the drug KH176 can effectively reduce elevated cellular levels of reactive oxygen species and protect primary cells deficient in oxidative phosphorylation from redox disorders [27,70]. |
| Ru360 | The study by Pool et al. demonstrated that Ru360 prevents mitochondrial overload of Ca2+, dysfunction of this organelle, and, consequently, contractile dysfunction. However, it is used only in preclinical settings [34]. |
| Antioxidant SS31 | The antioxidant SS31, currently tested in clinical trials, improves the coupling of electron transport chain complexes, and thus enhances mitochondrial bioenergetics and suppresses the abundance of ROS and oxidative stress [34]. |
| NAD+ supplementation | It is a possibility for preserving mitochondrial function since homeostasis of NAD+ improves function by reducing oxidative stress and DNA damage [34]. |
| L-glutamine | It has nutraceutical potential for the treatment of AF, as it stabilizes the microtubular network, increases the expression of heat shock protein in degenerative and inflammatory diseases, and contributes to the suppression of ROS and DNA damage induced by ROS due to its antioxidant activity [34]. |
| Therapeutic Possibilities | Main Effects |
|---|---|
| Statins | Reduction of C-reactive protein (CRP); prevention of inflammation, consequently preventing electrical and structural remodeling; prevention of oxygen free radical (ROS) synthesis induced by NADPH oxidase. |
| Steroids | Anti-inflammatory activity, indirect antioxidant, and immunomodulatory properties. Promotes reduction of atrial endothelial protein nitric oxide synthase levels and CRP levels. |
| Carvedilol | α1 blocking and antioxidant properties and anti-oxidation effects, in addition to exerting modulating effects on ionic channels and currents. |
| Dipeptidyl Peptidase-4 inhibitors | Reduction of ROS, promotion of mitochondrial oxidative stress, improvement of mitochondrial function, preservation of mitochondrial biogenesis, and reduction of inflammation. |
| Selective Sodium-Glucose Cotransporter 2 Inhibitors | Reduction of arterial resistance, improving endothelial function; normalization of sodium and calcium cytosolic concentrations; reduction of ROS synthesis; promotion of prevention of atrial remodeling and reduction of atrial fibrillation (AF) burden; promotion of less systemic inflammation; inhibition of atrial fibrosis and cardiomyocyte hypertrophy. In addition, it promotes a 19% reduction in AF in patients with diabetes, regardless of pre-existing AF or heart failure. In addition, they are suspected of promoting the reduction of pro-inflammatory molecules, increasing adiponectin, and suppressing inflammatory markers in the myocardium. |
| Ubiquinone | Anti-inflammatory antioxidant activity has a beneficial effect on mitochondrial function and significantly suppresses DNA damage. |
| Thiazolidinediones | Reduction of atrial remodeling. They prevent the recurrence of AF after electrical cardioversion, reduce cardiac risk factors and surrogate indicators of cardiovascular disease, and reduce the frequency of cardiac events in individuals with diabetes. |
| Trimetazidine | Reduction of ROS synthesis by acting directly on the activity of the respiratory chain. In addition, it prevents structural atrial remodeling, reduces the inducibility of AF, and shortens the duration of AF. |
| Ranolazine | Reduction of oxidative stress, improvement of mitochondrial function, suppression of apoptosis, and reduction of the likelihood of developing AF by approximately 50%. In addition, it increases the success rate of amiodarone cardioversion. |
| A diet rich in antioxidants | Vitamins E and C are antioxidants and eliminate ROS, such as O2, OH, peroxynitrite, sulfhydryl radicals, and oxidized low-density lipoprotein. |
| Mitochondrial transcription factor A (TFAM) | It increases ATP content by upregulating NADH-1 mitochondrial-coded dehydrogenase and cytochrome c oxidase-1 mitochondrially-coded expression levels. |
| Relaxin-2 | Reduction of oxidative stress (decrease in plasma levels of hydrogen peroxide and ROS), inhibition of profibrotic molecules, and suppression of inflammation, with a decrease in gene expression of inflammatory markers. In vitro, treatment with relaxin-2 inhibited the migration of normal human atrial cardiac fibroblasts. Furthermore, it reduced mRNA and protein levels of the profibrotic molecule, transforming growth factor-beta1 (TGF-β1). |
| Costunolide | Reduces inflammation and fibrosis induced by angiotensin II, improves mitochondrial function, alleviates oxidative stress by countering excessive ROS production, and activates the factor-2-related erythroid nuclear signaling pathway. |
| Febuxostat | Reduces the production of ROS, inhibits xanthine oxidase, and combats oxidative stress and inflammation, showing a decrease in inflammatory markers and the activity of antioxidant enzymes. Additionally, it positively influences AF by regulating the TGF-β1/Smad signaling pathway, which plays a role in collagen production and fibrosis. |
| Aliskiren | Attenuates electrical and structural atrial remodeling induced by rapid atrial pacing, reducing inflammation and oxidative stress. Furthermore, it regulates the PI3K/Akt signaling pathway. |
| Wenxin Keli | Antiarrhythmic properties and selective inhibition of atrial sodium current. It improves mitochondrial function by increasing respiration and reducing ROS production. In diabetic rats, Wenxin Keli prevents AF by enhancing atrial remodeling and restoring mitochondrial function. |
| Hydrogen sulfide | Activation of the PI3K/Akt/eNOS signaling pathway is associated with a reduction in the production of ROS. H2S can reduce diabetes-induced AF, decreasing the incidence and persistence of AF without affecting glucose metabolism. |
| Andrographolide | Reduction of cardiac cell apoptosis, improvement of mitochondrial function, antioxidant role, regulation of calcium homeostasis genes, and influence on transcription factors like factor-2-related erythroid nuclear. |
| Metformin | Activation of AMPK Src kinase, normalization of connective tissue expression, and prevention of atrial remodeling via the AMPK/PGC-1/PPAR pathway. Preserves mitochondrial function, improving the oxygenation and activity of complexes I, II, and IV. Increases PGC-1 and Coenzyme Q10 expression, providing antioxidant benefits and membrane stabilization. |
| Fibrates | Impact mitochondrial function through the PPAR/PGC-1 pathway, potentially mitigating metabolic remodeling by regulating the PPAR/sirtuin route 1/PGC-1, thereby reversing the shortening of the atrial refractory period. |
| Elamipretide | It improves mitochondrial efficiency and reduces the production of ROS by stabilizing the mitochondrial membrane and cytochrome C, increasing ATP production, normalizing the ATP/ADP ratio, and reducing TNF and CRP levels. |
| Genetic therapy | Restores average heart rate and improves heart rate control in animal models of AF. However, they have not yet reached the phase of widespread clinical use. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menezes Júnior, A.d.S.; França-e-Silva, A.L.G.d.; Oliveira, J.M.d.; Silva, D.M.d. Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review. Int. J. Mol. Sci. 2024, 25, 535. https://doi.org/10.3390/ijms25010535
Menezes Júnior AdS, França-e-Silva ALGd, Oliveira JMd, Silva DMd. Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review. International Journal of Molecular Sciences. 2024; 25(1):535. https://doi.org/10.3390/ijms25010535
Chicago/Turabian StyleMenezes Júnior, Antônio da Silva, Ana Luísa Guedes de França-e-Silva, Joyce Monteiro de Oliveira, and Daniela Melo da Silva. 2024. "Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review" International Journal of Molecular Sciences 25, no. 1: 535. https://doi.org/10.3390/ijms25010535
APA StyleMenezes Júnior, A. d. S., França-e-Silva, A. L. G. d., Oliveira, J. M. d., & Silva, D. M. d. (2024). Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review. International Journal of Molecular Sciences, 25(1), 535. https://doi.org/10.3390/ijms25010535