
Venetoclax and Hypomethylating Agent Combination in Myeloid Malignancies: Mechanisms of Synergy and Challenges of Resistance


Abstract
:1. Introduction
2. Mechanisms of Resistance against Monotherapies with Ven or HMA in AML Cell Lines
2.1. Resistance against Ven
2.2. Resistance against HMA
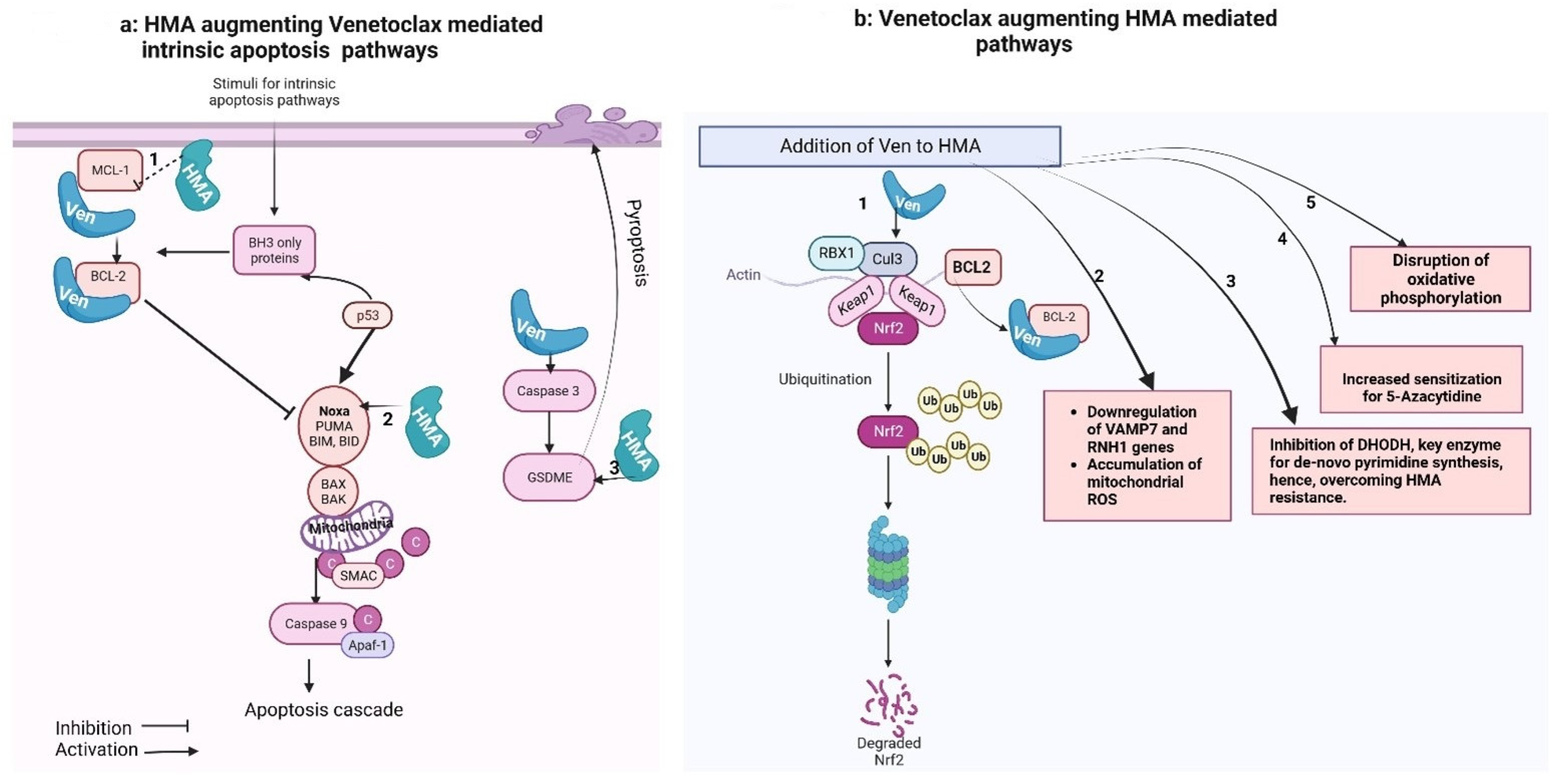
3. Ven–HMA Synergy Mechanisms
3.1. Preclinical Data
3.1.1. HMA-Mediated Downregulation of MCL-1 Levels
3.1.2. BCL2 Family Protein as 5-Aza-Sensitizing Targets
3.1.3. Combination Disrupts Energy Metabolism and Targets Leukemia Stem Cells (LSCs)
3.1.4. Reactive Oxygen Species (ROS)-Dependent Antileukemic Activity
3.1.5. HMA (5-Aza) Induced “Priming” of the AML Cells for Ven-Induced Apoptosis
3.1.6. Overexpression or HMA-Mediated Restoration of Caspase-3/GSDME Significantly Increases Ven-Induced Pyroptosis
3.1.7. Ven Augments HMA via Inhibiting De Novo Pyrimidine Synthesis

{kind=link}
| Azacytidine (5-Aza) Supports Venetoclax (Ven) | |
| Tsao et al. [32] | HMA mediated downregulation of MCL-1 5-Aza down-regulates MCL-1 in a p53-independent manner, leading to enhanced apoptosis in AML cells when combined with ABT-737 (inhibitor of BCL-2 and BCL-xL) |
| Jin et al. [41] | Priming of AML cells for Ven-induced apoptosis 5-Azacitidine induces “priming” through integrated stress response -mediated induction of PMAIP1 (gene for NOXA protein) transcripts, sensitizing AML cells to Ven-induced apoptosis |
| Ye et al. [46] | HMA mediated restoration of Caspase-3/GSDME significantly increases Ven-induced pyroptosis Venetoclax triggers pyroptosis in AML cells by cleaving GSDME, which is downregulated due to promoter methylation in AML cells and is associated with poor prognosis. GSDME overexpression, achieved through gene demethylation or HMA treatment, enhances Venetoclax-induced pyroptosis in AML. |
| Ven supports 5-Aza | |
| Bogenberger et al. [34] | BCL-2 family proteins in 5-Aza sensitization RNAi-mediated knockdown of BCL-xL sensitizes AML cells to 5-Aza, with BCL-xL and MCL-1 playing a crucial role in sensitization. Navitoclax (combined inhibitor of BCL-2, Bcl-xL and Bcl-w) was reportedly more potent than Ven (a selective BCL2 inhibitor) in enhancing 5-Aza activity, highlighting the significance of targeting BCL-xL, BCL-w, and BCL2 |
| Gu et al. [48] | Ven mediated inhibition of de-novo pyrimidine synthesis Upregulated de-novo pyrimidine synthesis, mediated by key mitochondrial enzyme DHODH, is the major adaptive resistance mechanism against HMA. Ven, by inhibiting BCL-2, depolarizes the mitochondrial membrane and hence inhibit the DHODH enzyme, leading to overcoming the resistance against HMA. |
| Novel mechanism with Ven + Aza combination | |
| Pollyea et al. [36] | Disruption of TCA cycle in leukemia stem cells Ven + 5-Aza disrupts the tricarboxylic acid cycle, targeting oxidative phosphorylation in leukemia stem cells |
| Nguyen et al. [30] Hu et al. [39] Kamachi et al. [40] | Reactive oxygen species-dependent mechanisms |
| |
3.2. Clinical Data: Molecular Predictors for Response to Ven–HMA Combination
| Study | Type of Study | Population | Favorable Predictors | Unfavorable Predictors | Key Findings |
|---|---|---|---|---|---|
| Stahl et al. [11] | Retrospective | 86 patients with relapsed/refractory AML | NPM1 gene mutations | Adverse cytogenetics, TP53, KRAS/NRAS, SF3B1 mutations | Higher response rates and OS with azacitidine + venetoclax. NPM1 mutations associated with better response. Adverse cytogenetics and mutations in TP53, KRAS/NRAS, SF3B1 associated with worse OS. |
| Morisa et al. [12] | Retrospective | 86 patients (newly diagnosed and relapsed/refractory AML) | CEPBA mutation | - | CEPBA mutation favored CR/CRi. Higher overall survival for complete responders compared to non-responders. |
| DiNardo et al. [15] | Clinical trial | 86 patients (44 newly diagnosed AML, 42 relapsed/refractory AML) frontline therapy in old patients with AML | NPM1 or IDH2 mutations | FLT3 or RAS or TP53 mutations | High response rates and durable remissions with NPM1 or IDH2 mutations. Primary and adaptive resistance characterized by FLT3, RAS, or TP53 mutations. |
| Johnson et al. [53] | Retrospective | relapsed/refractory AML | ASXL1 gene mutation | TP53 mutations, absence of IDH1/2 mutations, non-achievement of CR/CRi | ASXL1 gene mutation predicted superior response. TP53 mutations predicted inferior response. |
| Gangat et al. [54] | Retrospective | 103 patients treatment naïve AML | ASXL1 mutation, absence of TP53 and FLT3-ITD mutations | Presence of TP53 mutation | ASXL1 mutation and absence of TP53 and FLT3-ITD predicted favorable response. ASXL1 mutations and adverse karyotype predicted inferior survival. |
| Weng et al. [55] | Retrospective | 150 Chinese patient population with relapsed/refractory AML | Mutations in IDH1/2, NPM1, ASXL1, chromatin–cohesin genes | Adverse cytogenetics, mutations in FLT3-ITD, K/NRAS | Adverse cytogenetics and ELN adverse risk predicted inferior response. Mutations in IDH1/2, NPM1, ASXL1, and chromatin–cohesin genes predicted superior response. |
3.3. Superior Response with Ven–HMA Compared to Ven-LDAC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Key Statistics for Acute Myeloid Leukemia (AML). Available online: https://www.cancer.org/cancer/types/acute-myeloid-leukemia/about/key-statistics.html (accessed on 29 October 2023).
- FDA. FDA Grants Regular Approval to Venetoclax in Combination for Untreated Acute Myeloid Leukemia. 11 June 2021. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-venetoclax-combination-untreated-acute-myeloid-leukemia (accessed on 24 September 2023).
- Uy, N.; Singh, A.; Gore, S.D.; Prebet, T. Hypomethylating agents (HMA) treatment for myelodysplastic syndromes: Alternatives in the frontline and relapse settings. Expert Opin. Pharmacother. 2017, 18, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef] [PubMed]
- Šorm, F.; Pískala, A.; Čihák, A.; Veselý, J. 5-Azacytidine, a new, highly effective cancerostatic. Cell. Mol. Life Sci. 1964, 20, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Carraway, H.E.M. Overview of the Management of Higher-Risk Myelodysplastic Syndromes. Cancer J. 2023, 29, 160–167. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Stahl, M.; Menghrajani, K.; Derkach, A.; Chan, A.; Xiao, W.; Glass, J.; King, A.C.; Daniyan, A.F.; Famulare, C.; Cuello, B.M.; et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021, 5, 1552–1564. [Google Scholar] [CrossRef]
- Morsia, E.; McCullough, K.; Joshi, M.; Cook, J.; Alkhateeb, H.B.; Al-Kali, A.; Begna, K.; Elliott, M.; Hogan, W.; Litzow, M.; et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am. J. Hematol. 2020, 95, 1511–1521. [Google Scholar] [CrossRef]
- Pollyea, D.A.; DiNardo, C.D.; Arellano, M.L.; Pigneux, A.; Fiedler, W.; Konopleva, M.; Rizzieri, D.A.; Smith, B.D.; Shinagawa, A.; Lemoli, R.M.; et al. Impact of Venetoclax and Azacitidine in Treatment-Naïve Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 2753–2761. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bao, X.; Xue, S.-L.; Shen, H.; Cen, J.; Yao, L.; Pan, J.; Zhu, M.; Liu, D.; Hu, X.; et al. Venetoclax with decitabine as frontline treatment in younger adults with newly diagnosed ELN adverse-risk AML. Blood 2023, 142, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Ong, F.; Kim, K.; Konopleva, M.Y. Venetoclax resistance: Mechanistic insights and future strategies. Cancer Drug Resist. 2022, 5, 380–400. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, R.; Moison, C.; Thiollier, C.; Krosl, J.; Bordeleau, M.E.; Lehnertz, B.; Lavallée, V.P.; MacRae, T.; Mayotte, N.; Labelle, C.; et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2020, 34, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers Of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, E202–E205. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- Dumon, S.; Santos, S.; Debierre-Grockiego, F.; Gouilleux-Gruart, V.; Cocault, L.; Boucheron, C.; Mollat, P.; Gisselbrecht, S.; Gouilleux, F. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene 1999, 18, 4191–4199. [Google Scholar] [CrossRef]
- Kasper, S.; Breitenbuecher, F.; Heidel, F.; Hoffarth, S.; Markova, B.; Schuler, M.; Fischer, T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Kuusanmäki, H.; Leppä, A.-M.; Pölönen, P.; Kontro, M.; Dufva, O.; Deb, D.; Yadav, B.; Brück, O.; Kumar, A.; Everaus, H.; et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 2019, 105, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Geng, S.; Weng, J.; Deng, C.; Lu, Z.; Luo, C.; Du, X. The hENT1 and DCK genes underlie the decitabine response in patients with myelodysplastic syndrome. Leuk. Res. 2015, 39, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Masala, E.; Rossi, A.; Martino, A.; Sanna, A.; Buchi, F.; Canzian, F.; Cilloni, D.; Gaidano, V.; Voso, M.T.; et al. Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 2014, 28, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.; Franich, R.L.; Shortt, J.; Johnstone, R.W.; Kats, L.M. Distinct and overlapping mechanisms of resistance to azacytidine and guadecitabine in acute myeloid leukemia. Leukemia 2020, 34, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.-P.J. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 inhibitor venetoclax inhibits Nrf2 antioxidant pathway activation induced by hypomethylating agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef]
- Cheng, J.X.; Chen, L.; Li, Y.; Cloe, A.; Yue, M.; Wei, J.; Watanabe, K.A.; Shammo, J.M.; Anastasi, J.; Shen, Q.J.; et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018, 9, 1163, Erratum in Nat. Commun. 2018, 9, 2286. [Google Scholar] [CrossRef]
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Qiu, Y.H.; Zhang, N.; Coombes, K.R.; et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann. Hematol. 2012, 91, 1861–1870. [Google Scholar] [CrossRef]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Aguilar, L.C.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 inhibition with venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Bogenberger, J.M.; Kornblau, S.M.; E Pierceall, W.; Lena, R.; Chow, D.; Shi, C.-X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraju, D.V.; Alapa, M.; Polonskaia, A.; Risueño, A.; Subramanyam, P.; Anand, A.; Ghosh, K.; Kyriakopoulos, C.; Hemerich, D.; Hurren, R.; et al. Extended exposure to low doses of azacitidine induces differentiation of leukemic stem cells through activation of myeloperoxidase. Haematologica, 2023; Epub ahead of printing. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, L.; Nkwocha, J.; Sharma, K.; Zhou, L.; Grant, S. Synergistic Interactions between the Hypomethylating Agent Thio-Deoxycytidine and Venetoclax in Myelodysplastic Syndrome Cells. Hematol. Rep. 2023, 15, 91–100. [Google Scholar] [CrossRef]
- Kamachi, K.; Ureshino, H.; Watanabe, T.; Yoshida-Sakai, N.; Fukuda-Kurahashi, Y.; Kawasoe, K.; Hoshiko, T.; Yamamoto, Y.; Kurahashi, Y.; Kimura, S. Combination of a New Oral Demethylating Agent, OR2100, and Venetoclax for Treatment of Acute Myeloid Leukemia. Cancer Res. Commun. 2023, 3, 297–308. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef]
- Jonas, B.A.; Pollyea, D.A. How we use venetoclax with hypomethylating agents for the treatment of newly diagnosed patients with acute myeloid leukemia. Leukemia 2019, 33, 2795–2804. [Google Scholar] [CrossRef]
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.; Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert, E.R.; et al. Pevonedistat and azacitidine upregulate NOXA (PMAIP1) to increase sensitivity to venetoclax in preclinical models of acute myeloid leukemia. Haematologica 2021, 107, 825–835. [Google Scholar] [CrossRef]
- Nakajima, W.; Miyazaki, K.; Sakaguchi, M.; Asano, Y.; Ishibashi, M.; Kurita, T.; Yamaguchi, H.; Takei, H.; Tanaka, N. Epigenetic Priming with Decitabine Augments the Therapeutic Effect of Cisplatin on Triple-Negative Breast Cancer Cells through Induction of Proapoptotic Factor NOXA. Cancers 2022, 14, 248. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Hollingsworth, L.R.; Wu, H. Mechanism and Regulation of Gasdermin-Mediated Cell Death. Cold Spring Harb. Perspect. Biol. 2020, 12, a036400. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, W.; Fan, C.; Dong, J.; Peng, M.; Deng, W.; Zhang, H.; Yang, L. Antileukemic effect of venetoclax and hypomethylating agents via caspase-3/GSDME-mediated pyroptosis. J. Transl. Med. 2023, 21, 606. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 2020, 35, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Tohme, R.; Goldfinger, M.; Tomlinson, B.K.; Sakre, N.; Hanmer, S.; Jha, B.K.; Maciejewski, J.P.; Verma, A.; Saunthararajah, Y. Venetoclax Inhibition of Pyrimidine Synthesis Guides Methods for Integration with Decitabine or 5-Azacytidine That Are Non-Myelosuppressive. Blood 2020, 136 (Suppl. 1), 26–27. [Google Scholar] [CrossRef]
- Saunthararajah, Y. Mysteries of partial dihydroorotate dehydrogenase inhibition and leukemia terminal differentiation. Haematologica 2020, 105, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves Venetoclax in Combination for AML in Adults. 20 December 2019. Available online: https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults (accessed on 24 September 2023).
- Chyla, B.J.; Harb, J.; Mantis, C.; Riehm, J.J.; Ross, J.A.; Sun, Y.; Huang, X.; Jiang, Q.; Dail, M.; Peale, F.V., Jr.; et al. Response to Venetoclax in Combination with Low Intensity Therapy (LDAC or HMA) in Untreated Patients with Acute Myeloid Leukemia Patients with IDH, FLT3 and Other Mutations and Correlations with BCL2 Family Expression. Blood 2019, 134, 546. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Johnson, I.M.; Ilyas, R.; McCullough, K.; Al-Kali, A.; Alkhateeb, H.B.; Begna, K.; Mangaonkar, A.A.; Litzow, M.R.; Hogan, W.J.; Shah, M.V.; et al. Molecular Predictors of Response and Survival in Patients with Relapsed/Refractory Acute Myeloid Leukemia Following Venetoclax Plus Hypomethylating Agent Therapy. Blood 2022, 140 (Suppl. 1), 3233–3234. [Google Scholar] [CrossRef]
- Gangat, N.; Johnson, I.; McCullough, K.; Farrukh, F.; Al-Kali, A.; Alkhateeb, H.; Begna, K.; Mangaonkar, A.; Litzow, M.; Hogan, W.; et al. Molecular predictors of response to venetoclax plus hypomethylating agent in treatment-naïve acute myeloid leukemia. Haematologica 2022, 107, 2501–2505. [Google Scholar] [CrossRef]
- Weng, G.; Zhang, Y.; Yu, G.; Luo, T.; Yu, S.; Xu, N.; Sun, Z.; Lin, D.; Deng, L.; Liang, X.; et al. Genetic characteristics predict response to venetoclax plus hypomethylating agents in relapsed or refractory acute myeloid leukemia. J. Intern. Med. 2022, 293, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Sanber, K.; Ye, K.; Tsai, H.-L.; Newman, M.; Webster, J.A.; Gojo, I.; Ghiaur, G.; Prince, G.T.; Gondek, L.P.; Smith, B.D.; et al. Venetoclax in combination with hypomethylating agent for the treatment of advanced myeloproliferative neoplasms and acute myeloid leukemia with extramedullary disease. Leuk. Lymphoma 2023, 64, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.; Tadi, P. Cytarabine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK557680/ (accessed on 28 October 2023).
- Megías-Vericat, J.E.; Montesinos, P.; Herrero, M.J.; Bosó, V.; Martínez-Cuadrón, D.; Poveda, J.L.; Sanz, M.; Aliño, S.F. Pharmacogenomics and the treatment of acute myeloid leukemia. Pharmacogenomics 2016, 17, 1245–1272. [Google Scholar] [CrossRef]
| Genetic Mutation | Response to Ven–HMA Combination | Proposed or Reported Mechanism |
|---|---|---|
| NPM1 | Predicts positive response | Unclear mechanism. |
| IDH1/IDH2 | Predicts positive response | IDH1/2 mutations induce neomorphic enzyme activity, producing R-2-hydroxyglutarate that inhibits cytochrome c oxidase, leading to apoptosis activation through BAX and BAK. However, the anti-apoptotic gene BCL-2 could antagonize BAX and BAK, thereby preventing apoptosis [25]. The BCL-2 antagonism by Ven counteracts this, promoting leukemic cell death and may explain the positive response in IDH1/2 mutant AML with Ven–HMA therapy. [13] |
| KRAS | Predicts resistance | KRAS mutation causes upregulation of MCL-1 and BCL2A1, downregulates BCL-2 and BAX. Contributes to Venetoclax resistance by upregulating anti-apoptotic proteins [19]. |
| PTPN11 | Predicts resistance | PTPN11 mutation causes upregulation of MCL-1 and BCL-xL. Contributes to Venetoclax resistance by upregulating anti-apoptotic proteins [19]. |
| FLT3 | Predicts resistance | FLT3-ITD mutation confers Venetoclax resistance by upregulation of BCL-xL and MCL-1 through complex downstream pathways [21,22,23]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, R.; Zokaei Nikoo, M.; Veeraballi, S.; Singh, A. Venetoclax and Hypomethylating Agent Combination in Myeloid Malignancies: Mechanisms of Synergy and Challenges of Resistance. Int. J. Mol. Sci. 2024, 25, 484. https://doi.org/10.3390/ijms25010484
Mishra R, Zokaei Nikoo M, Veeraballi S, Singh A. Venetoclax and Hypomethylating Agent Combination in Myeloid Malignancies: Mechanisms of Synergy and Challenges of Resistance. International Journal of Molecular Sciences. 2024; 25(1):484. https://doi.org/10.3390/ijms25010484
Chicago/Turabian StyleMishra, Rahul, Maedeh Zokaei Nikoo, Sindhusha Veeraballi, and Abhay Singh. 2024. "Venetoclax and Hypomethylating Agent Combination in Myeloid Malignancies: Mechanisms of Synergy and Challenges of Resistance" International Journal of Molecular Sciences 25, no. 1: 484. https://doi.org/10.3390/ijms25010484
APA StyleMishra, R., Zokaei Nikoo, M., Veeraballi, S., & Singh, A. (2024). Venetoclax and Hypomethylating Agent Combination in Myeloid Malignancies: Mechanisms of Synergy and Challenges of Resistance. International Journal of Molecular Sciences, 25(1), 484. https://doi.org/10.3390/ijms25010484