
Glycolysis Reprogramming in Idiopathic Pulmonary Fibrosis: Unveiling the Mystery of Lactate in the Lung



Abstract
:1. Introduction
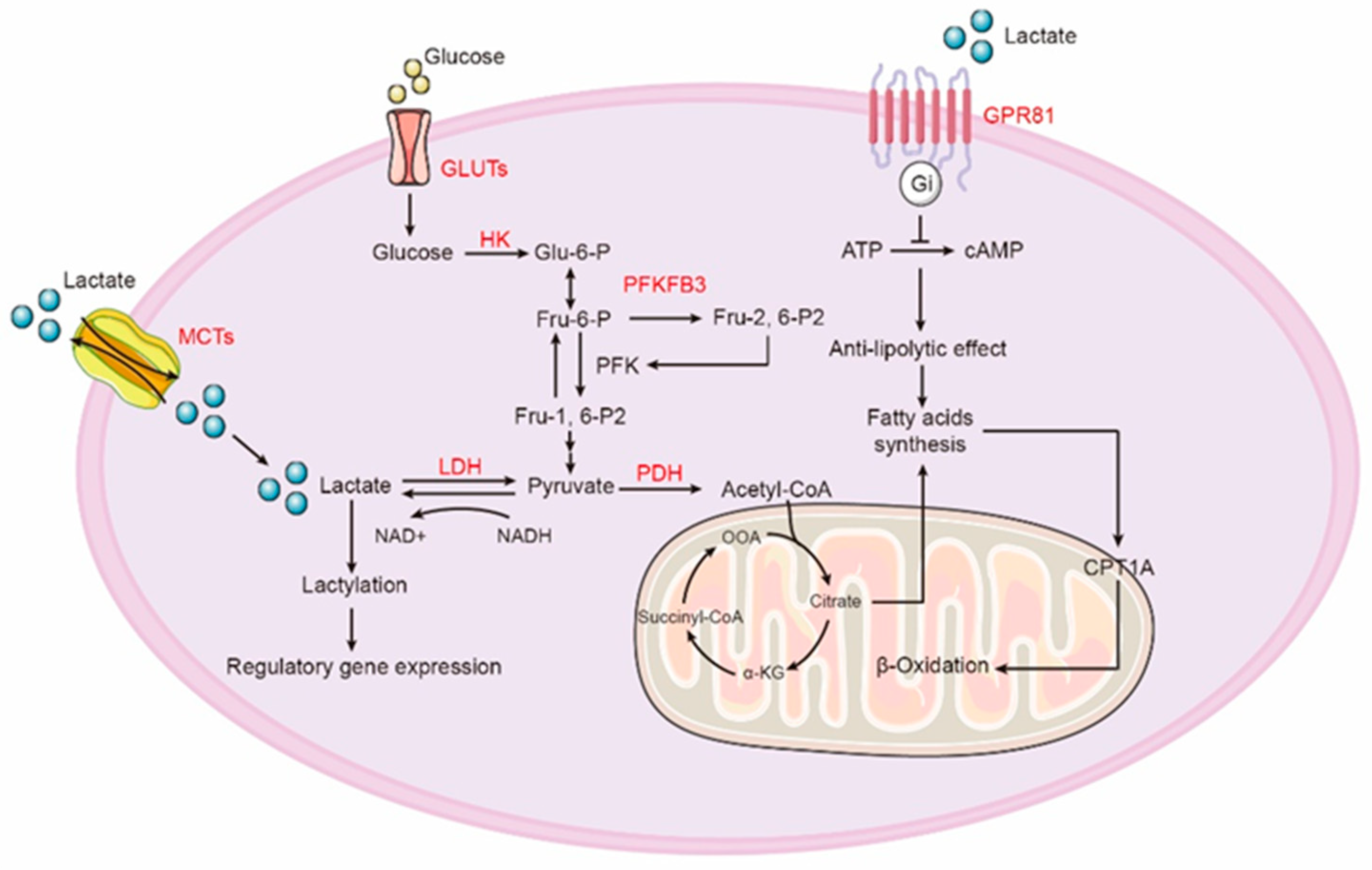
2. Aerobic Glycolysis
3. Glycolysis Reprogramming in Lung Fibrosis
4. Lactate Production and Oxidation
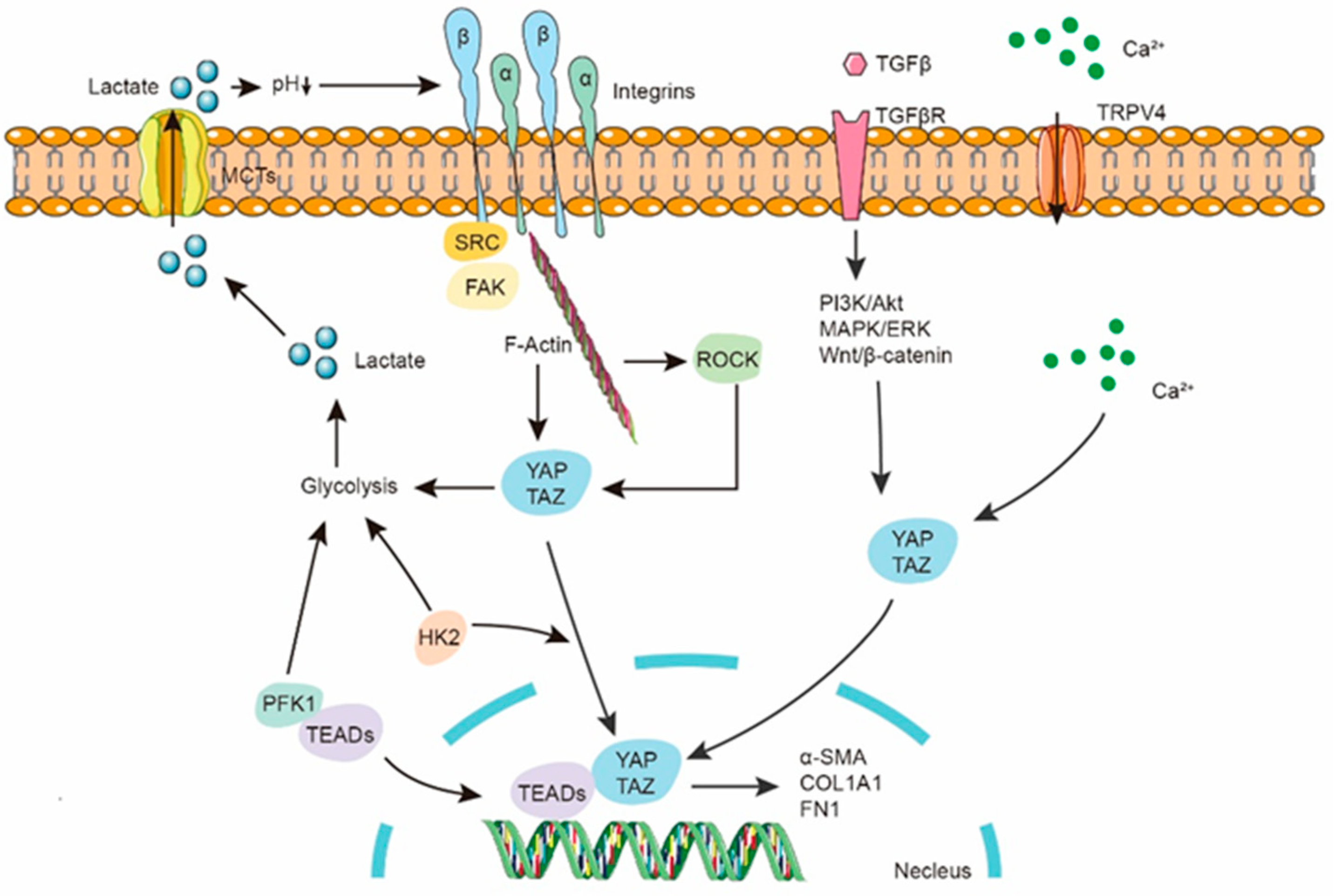
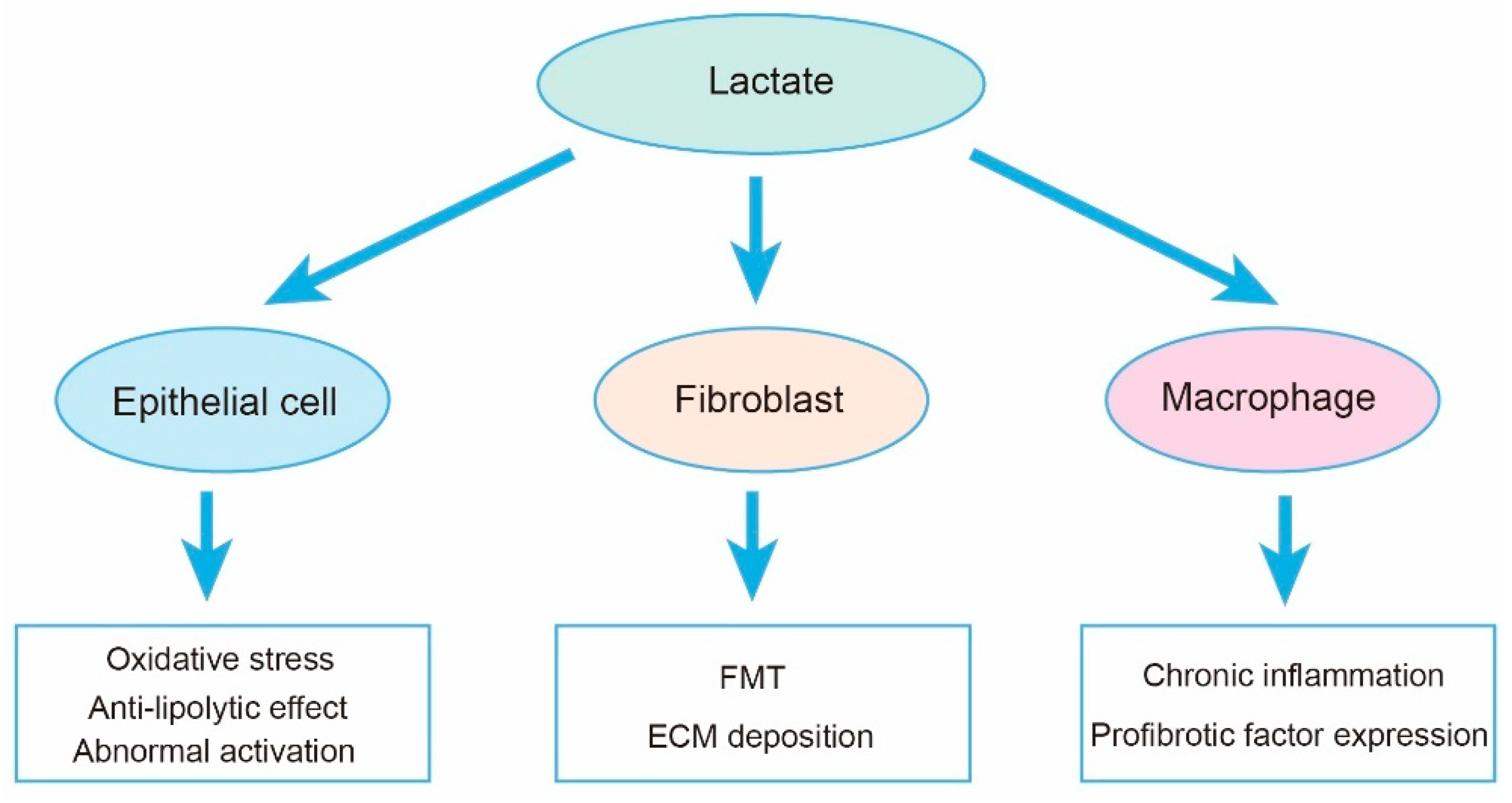
5. Lactate in Fibroblast
6. Lactate in AEC2s
7. Lactate in Inflammatory Cells
8. Lactate and Epigenetics
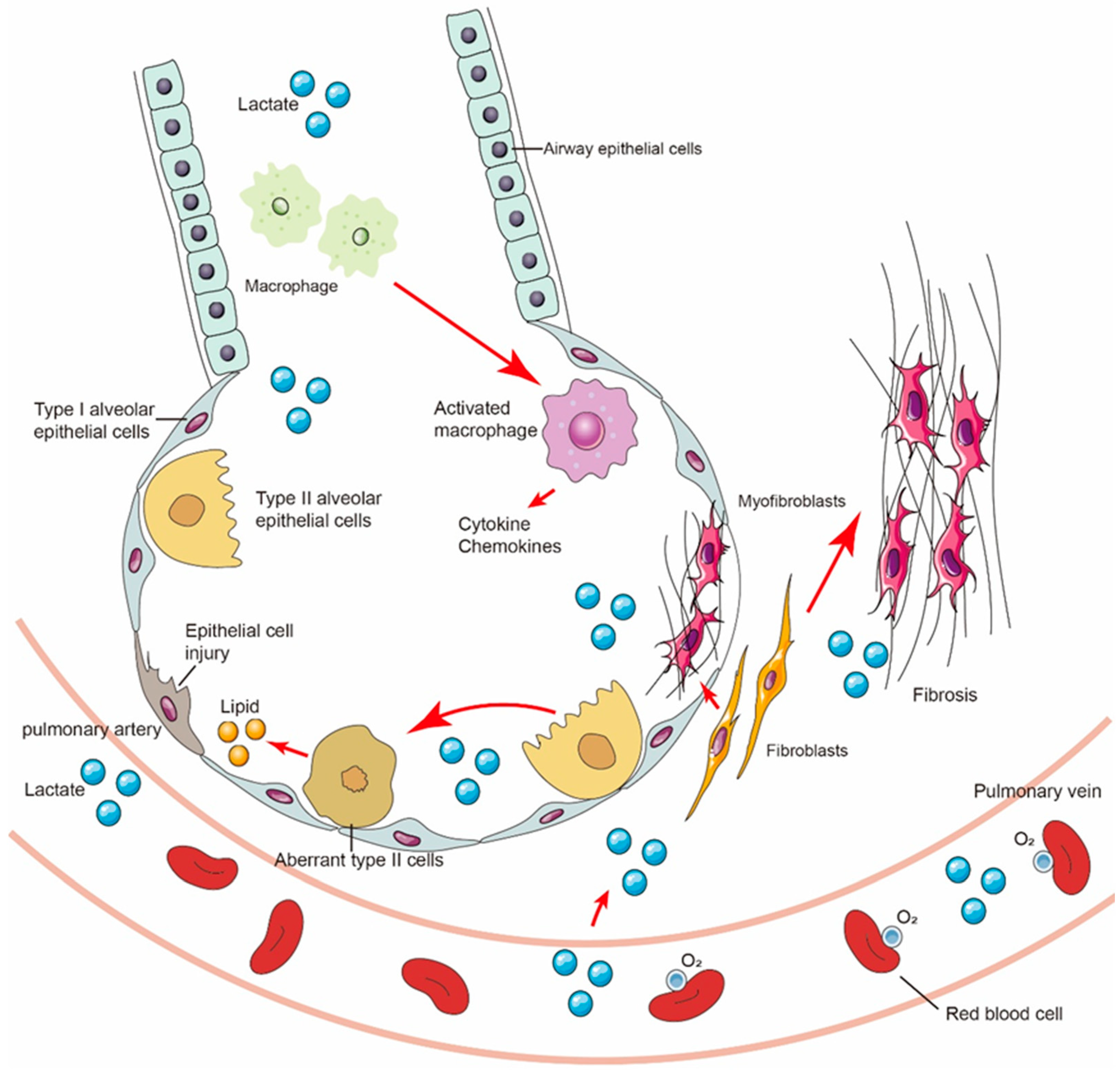
9. Lactate Shuttle and Multicellular Crosstalk in Fibrotic Lungs
10. Role of Lactate in Tissue Repair and Regeneration
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Picard, M. Why Do We Care More About Disease than Health? Phenomics 2022, 2, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Andersson, M.; Blanc, P.D.; Torén, K.; Järvholm, B. Smoking, occupational exposures, and idiopathic pulmonary fibrosis among Swedish construction workers. Am. J. Ind. Med. 2021, 64, 251–257. [Google Scholar] [CrossRef]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef]
- Ying, W. Phenomic Studies on Diseases: Potential and Challenges. Phenomics 2023, 3, 285–299. [Google Scholar] [CrossRef]
- Stowasser, S.; Hallmann, C. New guidelines for idiopathic pulmonary fibrosis. Lancet 2015, 386, 1823–1824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Refini, R.M.; D’alessandro, M.; Bergantini, L.; Cameli, P.; Vantaggiato, L.; Bini, L.; Landi, C. Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 5663. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Bhavsar, P.K.; Mumby, S.; Xu, B.; Hui, C.K.M.; Chung, K.F.; Adcock, I.M. Role of Metabolic Reprogramming in Pulmonary Innate Immunity and Its Impact on Lung Diseases. J. Innate Immun. 2020, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, L.; Zhang, W. Metabolic reprogramming: A novel metabolic model for pulmonary hypertension. Front. Cardiovasc. Med. 2022, 9, 957524. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Chu, S.G.; Villalba, J.A.; Liang, X.; Xiong, K.; Tsoyi, K.; Ith, B.; Ayaub, E.A.; Tatituri, R.V.; Byers, D.E.; Hsu, F.-F.; et al. Palmitic Acid-Rich High-Fat Diet Exacerbates Experimental Pulmonary Fibrosis by Modulating Endoplasmic Reticulum Stress. Am. J. Respir. Cell Mol. Biol. 2019, 61, 737–746. [Google Scholar] [CrossRef]
- Koudelka, A.; Cechova, V.; Rojas, M.; Mitash, N.; Bondonese, A.; Croix, C.S.; Ross, M.A.; Freeman, B.A. Fatty acid nitroalkene reversal of established lung fibrosis. Redox Biol. 2022, 50, 102226. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Y.; Liu, Q.; Mei, X.; Liu, J.; Tang, Y.; Luo, R.; Sun, D.; Ma, Y.; Wu, W.; et al. LDLR dysfunction induces LDL accumulation and promotes pulmonary fibrosis. Clin. Transl. Med. 2022, 12, e711. [Google Scholar] [CrossRef]
- Wang, L.; Yuan, H.; Li, W.; Yan, P.; Zhao, M.; Li, Z.; Zhao, H.; Wang, S.; Wan, R.; Li, Y.; et al. ACSS3 regulates the metabolic homeostasis of epithelial cells and alleviates pulmonary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1870, 166960. [Google Scholar] [CrossRef] [PubMed]
- Roque, W.; Romero, F. Cellular metabolomics of pulmonary fibrosis, from amino acids to lipids. Am. J. Physiol. Cell Physiol. 2021, 320, C689–C695. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhai, X.; Sun, X.; Cao, S.; Yuan, Q.; Wang, J. Metabolic reprogramming of pulmonary fibrosis. Front. Pharmacol. 2022, 13, 1031890. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, C.J.; Cottin, V.; Brown, K.K.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis: Shifting the paradigm. Eur. Respir. J. 2015, 46, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Teng, R.; Liu, Z.; Tang, H.; Zhang, W.; Chen, Y.; Xu, R.; Chen, L.; Song, J.; Liu, X.; Deng, H. HSP60 silencing promotes Warburg-like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol. 2019, 24, 101218. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef]
- Gopu, V.; Fan, L.; Shetty, R.S.; Nagaraja, M.R.; Shetty, S. Caveolin-1 scaffolding domain peptide regulates glucose metabolism in lung fibrosis. JCI Insight 2020, 5, e137969. [Google Scholar] [CrossRef]
- Umeda, Y.; Demura, Y.; Morikawa, M.; Anzai, M.; Kadowaki, M.; Ameshima, S.; Tsuchida, T.; Tsujikawa, T.; Kiyono, Y.; Okazawa, H.; et al. Prognostic Value of Dual-Time-Point 18F-FDG PET for Idiopathic Pulmonary Fibrosis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Pereira, K.M.A.; Chaves, F.N.; Viana, T.S.A.; Carvalho, F.S.R.; Costa, F.W.G.; Alves, A.P.N.N.; Sousa, F.B. Oxygen metabolism in oral cancer: HIF and GLUTs (Review). Oncol. Lett. 2013, 6, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta 2013, 1835, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Haneda, M.; Maeda, S.; Koya, D.; Kikkawa, R. TGF-beta 1 stimulates glucose uptake by enhancing GLUT1 expression in mesangial cells. Kidney Int. 1999, 55, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Hertenstein, H.; McMullen, E.; Weiler, A.; Volkenhoff, A.; Becker, H.M.; Schirmeier, S. Starvation-induced regulation of carbohydrate transport at the blood-brain barrier is TGF-β-signaling dependent. eLife 2021, 10, e62503. [Google Scholar] [CrossRef]
- Andrianifahanana, M.; Hernandez, D.M.; Yin, X.; Wang, Y.; Yi, E.S.; Roden, A.C.; Limper, A.H.; Leof, E.B.; Kang, J.-H.; Jung, M.-Y. Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J. 2016, 30, 3733–3744. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.S.; Lee, C.M.; Choi, A.M.; Stout-Delgado, H.W. Glucose Transporter 1-Dependent Glycolysis Is Increased during Aging-Related Lung Fibrosis, and Phloretin Inhibits Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 521–531. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.-S.; Nikahira, K.; Yun, H.S.; Harris, R.; Hong, K.S.; Huang, H.; Choi, A.M.K.; Stout-Delgado, H. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 2020, 75, 227–236. [Google Scholar] [CrossRef]
- Xie, N.; Cui, H.; Ge, J.; Banerjee, S.; Guo, S.; Dubey, S.; Abraham, E.; Liu, R.-M.; Liu, G. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L834–L844. [Google Scholar] [CrossRef]
- Mao, N.; Yang, H.; Yin, J.; Li, Y.; Jin, F.; Li, T.; Yang, X.; Sun, Y.; Liu, H.; Xu, H.; et al. Glycolytic Reprogramming in Silica-Induced Lung Macrophages and Silicosis Reversed by Ac-SDKP Treatment. Int. J. Mol. Sci. 2021, 22, 10063. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, B.; Azuelos, I.; Platé, M.; Guillotin, D.; Forty, E.J.; Contento, G.; Woodcock, H.V.; Redding, M.; Taylor, A.; Brunori, G.; et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β(1)-induced collagen biosynthesis. Sci. Signal. 2019, 12, eaav3048. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Cheng, D.; Li, G.; Liu, Y.; Li, P.; Sun, W.; Ma, D.; Ni, C. CircHIPK3 regulates pulmonary fibrosis by facilitating glycolysis in miR-30a-3p/FOXK2-dependent manner. Int. J. Biol. Sci. 2021, 17, 2294–2307. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Choudhury, M.; Kang, J.-H.; Schaefbauer, K.J.; Jung, M.-Y.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci. Signal. 2019, 12, eaax4067. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanni, G.T.; Han, W.; Sherrill, T.P.; Taylor, C.J.; Nichols, D.S.; Geis, N.M.; Singha, U.K.; Calvi, C.L.; McCall, A.S.; Dixon, M.M.; et al. Epithelial Yap/Taz are required for functional alveolar regeneration following acute lung injury. JCI Insight 2023, 8, e173374. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Tolosa, M.F.; Zhang, T.; Goru, S.K.; Severino, L.U.; Misra, P.S.; McEvoy, C.M.; Caldwell, L.; Szeto, S.G.; Gao, F.; et al. Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis. JCI Insight 2022, 7, e146243. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: Modulation, function and therapeutic approaches. Nat. Cancer 2023, 4, 9–26. [Google Scholar] [CrossRef]
- Philp, C.J.; Siebeke, I.; Clements, D.; Miller, S.; Habgood, A.; John, A.E.; Navaratnam, V.; Hubbard, R.B.; Jenkins, G.; Johnson, S.R. Extracellular Matrix Cross-Linking Enhances Fibroblast Growth and Protects against Matrix Proteolysis in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 594–603. [Google Scholar] [CrossRef]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.-E.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, Y.; Zhao, M.; Ding, L.; Yang, X.; Jing, Y.; Song, Y.; Chen, S.; Hu, Q.; Ni, Y. ITGB2-mediated metabolic switch in CAFs promotes OSCC proliferation by oxidation of NADH in mitochondrial oxidative phosphorylation system. Theranostics 2020, 10, 12044–12059. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Yaqoob, U.; Gan, C.; Jalan-Sakrikar, N.; Kostallari, E.; Lu, J.; Gao, J.; Sun, L.; Liu, M.; Sehrawat, T.S.; et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J. Hepatol. 2022, 77, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Xu, Q.; Hu, Y.; Tang, R.; Feng, J.; Zhou, Y.; Xing, S.; Gao, Y.; He, Z. Integrin β3-PKM2 pathway-mediated aerobic glycolysis contributes to mechanical ventilation-induced pulmonary fibrosis. Theranostics 2022, 12, 6057–6068. [Google Scholar] [CrossRef] [PubMed]
- Ata, R.; Antonescu, C.N. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int. J. Mol. Sci. 2017, 18, 189. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Kashihara, T.; Mukai, R.; Oka, S.-I.; Zhai, P.; Nakada, Y.; Yang, Z.; Mizushima, W.; Nakahara, T.; Warren, J.S.; Abdellatif, M.; et al. YAP mediates compensatory cardiac hypertrophy through aerobic glycolysis in response to pressure overload. J. Clin. Investig. 2022, 132, e150595. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef]
- Feng, Y.; Zou, R.; Zhang, X.; Shen, M.; Chen, X.; Wang, J.; Niu, W.; Yuan, Y.; Yuan, F. YAP promotes ocular neovascularization by modifying PFKFB3-driven endothelial glycolysis. Angiogenesis 2021, 24, 489–504. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, L.-F.; Miao, Y.; Xi, Y.; Li, X.; Liu, M.-F.; Zhang, M.; Li, B. Verteporfin Suppresses YAP-Induced Glycolysis in Breast Cancer Cells. J. Investig. Surg. Off. J. Acad. Surg. Res. 2023, 36, 2266732. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, J.; Zhao, X.; Wu, T.; Huang, Z.; Chen, D.; Liu, Y.; Ouyang, G. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep. 2020, 30, 793–806.e6. [Google Scholar] [CrossRef] [PubMed]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Neary, C.L.; Pastorino, J.G. Nucleocytoplasmic shuttling of hexokinase II in a cancer cell. Biochem. Biophys. Res. Commun. 2010, 394, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.-M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef]
- Xu, Q.; Mei, S.; Nie, F.; Zhang, Z.; Feng, J.; Zhang, J.; Qian, X.; Gao, Y.; He, Z.; Xing, S. The role of macrophage-fibroblast interaction in lipopolysaccharide-induced pulmonary fibrosis: An acceleration in lung fibroblast aerobic glycolysis. Lab. Investig. 2022, 102, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef]
- Bendahan, D.; Chatel, B.; Jue, T. Comparative NMR and NIRS analysis of oxygen-dependent metabolism in exercising finger flexor muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R740–R753. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic Acid: No Longer an Inert and End-Product of Glycolysis. Physiology 2017, 32, 453–463. [Google Scholar] [CrossRef]
- Yang, C.; Pan, R.Y.; Guan, F.; Yuan, Z. Lactate metabolism in neurodegenerative diseases. Neural Regen. Res. 2024, 19, 69–74. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Song, G.J.; Lee, M.G.; Jeoung, N.H.; Go, Y.; Harris, R.A.; Park, D.H.; Kook, H.; Lee, I.-K.; Suk, K. Metabolic Connection of Inflammatory Pain: Pivotal Role of a Pyruvate Dehydrogenase Kinase-Pyruvate Dehydrogenase-Lactic Acid Axis. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 14353–14369. [Google Scholar] [CrossRef] [PubMed]
- Soreze, Y.; Boutron, A.; Habarou, F.; Barnerias, C.; Nonnenmacher, L.; Delpech, H.; Mamoune, A.; Chrétien, D.; Hubert, L.; Bole-Feysot, C.; et al. Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J. Rare Dis. 2013, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Osmond, A.D.; Arevalo, J.A.; Duong, J.J.; Curl, C.C.; Moreno-Santillan, D.D.; Leija, R.G. Lactate as a myokine and exerkine: Drivers and signals of physiology and metabolism. J. Appl. Physiol. 2023, 134, 529–548. [Google Scholar] [CrossRef] [PubMed]
- Adepu, K.K.; Bhandari, D.; Anishkin, A.; Adams, S.H.; Chintapalli, S.V. Myoglobin Interaction with Lactate Rapidly Releases Oxygen: Studies on Binding Thermodynamics, Spectroscopy, and Oxygen Kinetics. Int. J. Mol. Sci. 2022, 23, 4747. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Tierney, D.F. Lactate metabolism in rat lung tissue. Arch. Intern. Med. 1971, 127, 858–860. [Google Scholar] [CrossRef]
- O’Neil, J.J.; Tierney, D.F. Rat lung metabolism: Glucose utilization by isolated perfused lungs and tissue slices. Am. J. Physiol. 1974, 226, 867–873. [Google Scholar] [CrossRef]
- Lottes, R.G.; Newton, D.A.; Spyropoulos, D.D.; Baatz, J.E. Lactate as substrate for mitochondrial respiration in alveolar epithelial type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L953–L961. [Google Scholar] [CrossRef]
- Rhoades, R.A.; Shaw, M.E.; Eskew, M.L.; Wali, S. Lactate metabolism in perfused rat lung. Am. J. Physiol. 1978, 235, E619–E623. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Routsi, C.; Bardouniotou, H.; Delivoria-Ioannidou, V.; Kazi, D.; Roussos, C.; Zakynthinos, S. Pulmonary lactate release in patients with acute lung injury is not attributable to lung tissue hypoxia. Crit. Care Med. 1999, 27, 2469–2473. [Google Scholar] [CrossRef]
- Mizock, B.A. Lung injury and lactate production: A hypoxic stimulus? Crit. Care Med. 1999, 27, 2585–2586. [Google Scholar] [CrossRef]
- Hu, X.; Xu, Q.; Wan, H.; Hu, Y.; Xing, S.; Yang, H.; Gao, Y.; He, Z. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Investig. 2020, 100, 801–811. [Google Scholar] [CrossRef]
- Lee, J.Y.; Stevens, R.P.; Kash, M.; Zhou, C.; Koloteva, A.; Renema, P.; Paudel, S.S.; Stevens, T. KD025 Shifts Pulmonary Endothelial Cell Bioenergetics and Decreases Baseline Lung Permeability. Am. J. Respir. Cell Mol. Biol. 2020, 63, 519–530. [Google Scholar] [CrossRef]
- Goodwin, J.; Choi, H.; Hsieh, M.-H.; Neugent, M.L.; Ahn, J.-M.; Hayenga, H.N.; Singh, P.K.; Shackelford, D.B.; Lee, I.-K.; Shulaev, V.; et al. Targeting Hypoxia-Inducible Factor-1α/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 216–231. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Jiang, Q.; Liang, Q.; Zhang, F.; Li, S.; Zhang, R.; Luan, J.; Zhu, J.; Gu, X.; et al. PKM2 promotes pulmonary fibrosis by stabilizing TGF-β1 receptor I and enhancing TGF-β1 signaling. Sci. Adv. 2022, 8, eabo0987. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Trawick, E.; Judge, J.L.; Wahl, L.A.; Epa, A.P.; Owens, K.M.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J.; O’Dwyer, D.N.; et al. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1305–L1312. [Google Scholar] [CrossRef]
- Judge, J.L.; Lacy, S.H.; Ku, W.-Y.; Owens, K.M.; Hernady, E.; Thatcher, T.H.; Williams, J.P.; Phipps, R.P.; Sime, P.J.; Kottmann, R.M. The Lactate Dehydrogenase Inhibitor Gossypol Inhibits Radiation-Induced Pulmonary Fibrosis. Radiat. Res. 2017, 188, 35–43. [Google Scholar] [CrossRef]
- Judge, J.L.; Nagel, D.J.; Owens, K.M.; Rackow, A.; Phipps, R.P.; Sime, P.J.; Kottmann, R.M. Prevention and treatment of bleomycin-induced pulmonary fibrosis with the lactate dehydrogenase inhibitor gossypol. PLoS ONE 2018, 13, e0197936. [Google Scholar] [CrossRef]
- Kang, Y.P.; Lee, S.B.; Lee, J.-M.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; Choi, C.W.; Shin, H.K.; Kim, D.-J.; Koh, E.S.; et al. Metabolic Profiling Regarding Pathogenesis of Idiopathic Pulmonary Fibrosis. J. Proteome Res. 2016, 15, 1717–1724. [Google Scholar] [CrossRef]
- Zhao, Y.D.; Yin, L.; Archer, S.; Lu, C.; Zhao, G.; Yao, Y.; Wu, L.; Hsin, M.; Waddell, T.K.; Keshavjee, S.; et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: A metabolomic study. BMJ Open Respir. Res. 2017, 4, e000183. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J.; Zhong, W.; Liu, Y.; Lu, Y.; Zeng, Z.; Huang, H.; Wan, X.; Meng, X.; Zou, F.; et al. Anlotinib Inhibits PFKFB3-Driven Glycolysis in Myofibroblasts to Reverse Pulmonary Fibrosis. Front. Pharmacol. 2021, 12, 744826. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef]
- Drent, M.; Cobben, N.A.; Henderson, R.F.; Wouters, E.F.; van Dieijen-Visser, M. Usefulness of lactate dehydrogenase and its isoenzymes as indicators of lung damage or inflammation. Eur. Respir. J. 1996, 9, 1736–1742. [Google Scholar] [CrossRef]
- Khan, A.A.; Allemailem, K.S.; Alhumaydhi, F.A.; Gowder, S.J.T.; Rahmani, A.H. The Biochemical and Clinical Perspectives of Lactate Dehydrogenase: An Enzyme of Active Metabolism. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 855–868. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Brereton, C.J.; Yao, L.; Davies, E.R.; Zhou, Y.; Vukmirovic, M.; Bell, J.A.; Wang, S.; Ridley, R.A.; Dean, L.S.N.; Andriotis, O.G.; et al. Pseudohypoxic HIF pathway activation dysregulates collagen structure-function in human lung fibrosis. eLife 2022, 11, e69348. [Google Scholar] [CrossRef]
- Zhu, Y.; Tan, J.; Xie, H.; Wang, J.; Meng, X.; Wang, R. HIF-1α regulates EMT via the Snail and β-catenin pathways in paraquat poisoning-induced early pulmonary fibrosis. J. Cell. Mol. Med. 2016, 20, 688–697. [Google Scholar] [CrossRef]
- de Peyster, A.; Wang, Y.Y. Genetic toxicity studies of gossypol. Mutat. Res. 1993, 297, 293–312. [Google Scholar] [CrossRef]
- Gadelha, I.C.; Fonseca, N.B.; Oloris, S.C.; Melo, M.M.; Soto-Blanco, B. Gossypol toxicity from cottonseed products. Sci. World J. 2014, 2014, 231635. [Google Scholar] [CrossRef]
- Di Stefano, G.; Manerba, M.; Di Ianni, L.; Fiume, L. Lactate dehydrogenase inhibition: Exploring possible applications beyond cancer treatment. Future Med. Chem. 2016, 8, 713–725. [Google Scholar] [CrossRef]
- Schruf, E.; Schroeder, V.; Kuttruff, C.A.; Weigle, S.; Krell, M.; Benz, M.; Bretschneider, T.; Holweg, A.; Schuler, M.; Frick, M.; et al. Human lung fibroblast-to-myofibroblast transformation is not driven by an LDH5-dependent metabolic shift towards aerobic glycolysis. Respir. Res. 2019, 20, 87. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; He, C.; Carter, A.B. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020, 33, 101426. [Google Scholar] [CrossRef]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef]
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008, 582, 3569–3576. [Google Scholar] [CrossRef]
- Rao, X.; Zhou, D.; Deng, H.; Chen, Y.; Wang, J.; Zhou, X.; Jie, X.; Xu, Y.; Wu, Z.; Wang, G.; et al. Activation of NLRP3 inflammasome in lung epithelial cells triggers radiation-induced lung injury. Respir. Res. 2023, 24, 25. [Google Scholar] [CrossRef]
- Newton, D.A.; Lottes, R.G.; Ryan, R.M.; Spyropoulos, D.D.; Baatz, J.E. Dysfunctional lactate metabolism in human alveolar type II cells from idiopathic pulmonary fibrosis lung explant tissue. Respir. Res. 2021, 22, 278. [Google Scholar] [CrossRef]
- Valdivieso, G.; Clauzure, M.; Massip-Copiz, M.M.; Cancio, C.E.; Asensio, C.J.A.; Mori, C.; Santa-Coloma, T.A. Impairment of CFTR activity in cultured epithelial cells upregulates the expression and activity of LDH resulting in lactic acid hypersecretion. Cell. Mol. Life Sci. 2019, 76, 1579–1593. [Google Scholar] [CrossRef]
- Massip-Copiz, M.M.; Valdivieso, G.; Clauzure, M.; Mori, C.; Asensio, C.J.A.; Aguilar, M.; Santa-Coloma, T.A. Epidermal growth factor receptor activity upregulates lactate dehydrogenase A expression, lactate dehydrogenase activity, and lactate secretion in cultured IB3-1 cystic fibrosis lung epithelial cells. Biochem. Cell Biol. 2021, 99, 476–487. [Google Scholar] [CrossRef]
- Patgiri, A.; Skinner, O.S.; Miyazaki, Y.; Schleifer, G.; Marutani, E.; Shah, H.; Sharma, R.; Goodman, R.P.; To, T.-L.; Bao, X.R.; et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat. Biotechnol. 2020, 38, 309–313. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Dey, T.; Liu, R.-M.; Antony, V.B.; Sanders, Y.Y.; Adams, T.S.; Gomez, J.L.; Thannickal, V.J.; et al. CD38 Mediates Lung Fibrosis by Promoting Alveolar Epithelial Cell Aging. Am. J. Respir. Crit. Care Med. 2022, 206, 459–475. [Google Scholar] [CrossRef]
- Oh, G.-S.; Lee, S.-B.; Karna, A.; Kim, H.-J.; Shen, A.; Pandit, A.; Lee, S.; Yang, S.-H.; So, H.-S. Cellular NAD(+) Level through NQO1 Enzymatic Action Has Protective Effects on Bleomycin-Induced Lung Fibrosis in Mice. Tuberc. Respir. Dis. 2016, 79, 257–266. [Google Scholar] [CrossRef]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Vlassenko, A.G.; Rundle, M.M.; Raichle, M.E. Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proc. Natl. Acad. Sci. USA 2004, 101, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Weidemann, M.J. Role and regulation of glucose metabolism in proliferating cells. J. Natl. Cancer Inst. 1979, 62, 3–8. [Google Scholar] [PubMed]
- Rabinowitz, J.D.; Enerbäck, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Nociti, V.; Petzold, A.; Gasperini, C.; Quartuccio, E.; Lazzarino, G.; Di Pietro, V.; Belli, A.; Signoretti, S.; Vagnozzi, R.; et al. Serum lactate as a novel potential biomarker in multiple sclerosis. Biochim. Biophys. Acta 2014, 1842, 1137–1143. [Google Scholar] [CrossRef]
- Kirwan, J.R. Synovial fluid lactate in septic arthritis. Lancet 1982, 1, 457. [Google Scholar] [CrossRef]
- Fujii, W.; Kawahito, Y.; Nagahara, H.; Kukida, Y.; Seno, T.; Yamamoto, A.; Kohno, M.; Oda, R.; Taniguchi, D.; Fujiwara, H.; et al. Monocarboxylate transporter 4, associated with the acidification of synovial fluid, is a novel therapeutic target for inflammatory arthritis. Arthritis Rheumatol. 2015, 67, 2888–2896. [Google Scholar] [CrossRef]
- Shantha GP, S.; Wasserman, B.; Astor, B.C.; Coresh, J.; Brancati, F.; Sharrett, A.R.; Young, J.H. Association of blood lactate with carotid atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Carotid MRI Study. Atherosclerosis 2013, 228, 249–255. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Raghu, G.; Brown, K.K.; Costabel, U.; Cottin, V.; du Bois, R.M.; Lasky, J.A.; Thomeer, M.; Utz, J.P.; Khandker, R.K.; McDermott, L.; et al. Treatment of idiopathic pulmonary fibrosis with etanercept: An exploratory, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2008, 178, 948–955. [Google Scholar] [CrossRef]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.-F.; Feichtinger, J.; Zelger, B.; Sgonc, R.; Wolfram, D. The immunology of fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Nielsen, B.U.; Kolpen, M.; Jensen, P.; Katzenstein, T.; Pressler, T.; Ritz, C.; Mathiesen, I.H.M.; Faurholt-Jepsen, D. Neutrophil count in sputum is associated with increased sputum glucose and sputum L-lactate in cystic fibrosis. PLoS ONE 2020, 15, e0238524. [Google Scholar] [CrossRef]
- Worlitzsch, D.; Meyer, K.C.; Döring, G. Lactate levels in airways of patients with cystic fibrosis and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 111. [Google Scholar] [CrossRef]
- Svedberg, F.R.; Brown, S.L.; Krauss, M.Z.; Campbell, L.; Sharpe, C.; Clausen, M.; Howell, G.J.; Clark, H.; Madsen, J.; Evans, C.M.; et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat. Immunol. 2019, 20, 571–580. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, D.; Li, Y.; Yang, F.; Hou, L.; Yu, Y.; Sun, C.; Duan, G.; Meng, C.; Yan, H.; et al. Paraquat promotes acute lung injury in rats by regulating alveolar macrophage polarization through glycolysis. Ecotoxicol. Environ. Saf. 2021, 223, 112571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, S.; Lindenthal, L.; Zeitler, L.; Behnke, K.; Schlösser, D.; Strobl, B.; Scheller, J.; El Kasmi, K.C.; Murray, P.J. Lactate and IL6 define separable paths of inflammatory metabolic adaptation. Sci. Adv. 2021, 7, eabg3505. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, C.; Ma, J.; Peng, P.; Ren, X.; Cai, S.; Shen, X.; Wu, Y.; Zhang, S.; Wang, X.; et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023, 5, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Di, C.; Chang, P.; Li, L.; Feng, Z.; Xiao, S.; Yan, X.; Xu, X.; Li, H.; Qi, R.; et al. Lactylated Histone H3K18 as a Potential Biomarker for the Diagnosis and Predicting the Severity of Septic Shock. Front. Immunol. 2021, 12, 786666. [Google Scholar] [CrossRef] [PubMed]
- Irizarry-Caro, R.A.; McDaniel, M.M.; Overcast, G.R.; Jain, V.G.; Troutman, T.D.; Pasare, C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc. Natl. Acad. Sci. USA 2020, 117, 30628–30638. [Google Scholar] [CrossRef] [PubMed]
- Lopez Krol, A.; Nehring, H.P.; Krause, F.F.; Wempe, A.; Raifer, H.; Nist, A.; Stiewe, T.; Bertrams, W.; Schmeck, B.; Luu, M.; et al. Lactate induces metabolic and epigenetic reprogramming of pro-inflammatory Th17 cells. EMBO Rep. 2022, 23, e54685. [Google Scholar] [CrossRef]
- Troutman, T.D.; Hu, W.; Fulenchek, S.; Yamazaki, T.; Kurosaki, T.; Bazan, J.F.; Pasare, C. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 2012, 109, 273–278. [Google Scholar] [CrossRef]
- Matsumura, T.; Oyama, M.; Kozuka-Hata, H.; Ishikawa, K.; Inoue, T.; Muta, T.; Semba, K.; Inoue, J.-I. Identification of BCAP-(L) as a negative regulator of the TLR signaling-induced production of IL-6 and IL-10 in macrophages by tyrosine phosphoproteomics. Biochem. Biophys. Res. Commun. 2010, 400, 265–270. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhai, Z.; Duan, J.; Wang, X.; Zhong, J.; Wu, L.; Li, A.; Cao, M.; Wu, Y.; Shi, H.; et al. Lactate: The Mediator of Metabolism and Immunosuppression. Front. Endocrinol. 2022, 13, 901495. [Google Scholar] [CrossRef]
- Chen, L.; Huang, L.; Gu, Y.; Cang, W.; Sun, P.; Xiang, Y. Lactate-Lactylation Hands between Metabolic Reprogramming and Immunosuppression. Int. J. Mol. Sci. 2022, 23, 11943. [Google Scholar] [CrossRef]
- Wang, J.; Yang, P.; Yu, T.; Gao, M.; Liu, D.; Zhang, J.; Lu, C.; Chen, X.; Zhang, X.; Liu, Y. Lactylation of PKM2 Suppresses Inflammatory Metabolic Adaptation in Pro-inflammatory Macrophages. Int. J. Biol. Sci. 2022, 18, 6210–6225. [Google Scholar] [CrossRef]
- Fan, M.; Yang, K.; Wang, X.; Chen, L.; Gill, P.S.; Ha, T.; Liu, L.; Lewis, N.H.; Williams, D.L.; Li, C. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci. Adv. 2023, 9, eadc9465. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Jiang, D.; Dey, T.; Matthews, Q.L.; Liu, R.-M.; Liu, G. Lung Myofibroblasts Promote Macrophage Profibrotic Activity through Lactate-induced Histone Lactylation. Am. J. Respir. Cell Mol. Biol. 2021, 64, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xie, D.; Xiao, T.; Cheng, C.; Wang, D.; Sun, J.; Wu, M.; Yang, Y.; Zhang, A.; Liu, Q. H3K18 lactylation promotes the progression of arsenite-related idiopathic pulmonary fibrosis via YTHDF1/m6A/NREP. J. Hazard. Mater. 2024, 461, 132582. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Lactate shuttles in nature. Biochem. Soc. Trans. 2002, 30, 258–264. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Brooks, G.A. Energy Flux, Lactate Shuttling, Mitochondrial Dynamics, and Hypoxia. Adv. Exp. Med. Biol. 2016, 903, 439–455. [Google Scholar]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Monocarboxylic acid transport. Compr. Physiol. 2013, 3, 1611–1643. [Google Scholar] [PubMed]
- Lee, G.H.; Kim, D.S.; Chung, M.J.; Chae, S.W.; Kim, H.R.; Chae, H.J. Lysyl oxidase-like-1 enhances lung metastasis when lactate accumulation and monocarboxylate transporter expression are involved. Oncol. Lett. 2011, 2, 831–838. [Google Scholar] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Stüwe, L.; Müller, M.; Fabian, A.; Waning, J.; Mally, S.; Noël, J.; Schwab, A.; Stock, C. pH dependence of melanoma cell migration: Protons extruded by NHE1 dominate protons of the bulk solution. J. Physiol. 2007, 585, 351–360. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Cupeiro, R.; Pérez-Prieto, R.; Amigo, T.; Gortázar, P.; Redondo, C.; González-Lamuño, D. Role of the monocarboxylate transporter MCT1 in the uptake of lactate during active recovery. Eur. J. Appl. Physiol. 2016, 116, 1005–1010. [Google Scholar] [CrossRef]
- Pellerin, L.; Connes, P.; Bisbal, C.; Lambert, K. Editorial: Lactate as a Major Signaling Molecule for Homeostasis. Front. Physiol. 2022, 13, 910567. [Google Scholar] [CrossRef]
- Johnson, M.L.; Hussien, R.; Horning, M.A.; Brooks, G.A. Transpulmonary pyruvate kinetics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R769–R774. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.; Newsholme, P.; Brennan, L.; Maechler, P. The importance of redox shuttles to pancreatic beta-cell energy metabolism and function. Biochem. Soc. Trans. 2006, 34, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N.; Tapia-Paez, I.; Ullah, M.S.; Parton, L.E.; Schuit, F.; Quintens, R.; Sipila, I.; Mayatepe, E.; et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qin, H.; Zhang, L.; Jia, C.; Chao, Z.; Qin, X.; Zhang, H.; Chen, C. Hyperoxia induces glucose metabolism reprogramming and intracellular acidification by suppressing MYC/MCT1 axis in lung cancer. Redox Biol. 2023, 61, 102647. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, L.; Chen, W.; Chen, Q.; Sun, J.; Wang, G. Novel strategies to improve tumour therapy by targeting the proteins MCT1, MCT4 and LAT1. Eur. J. Med. Chem. 2021, 226, 113806. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Philp, A.; Macdonald, A.L.; Watt, P.W. Lactate—A signal coordinating cell and systemic function. J. Exp. Biol. 2005, 208, 4561–4575. [Google Scholar] [CrossRef]
- Ge, H.; Weiszmann, J.; Reagan, J.D.; Gupte, J.; Baribault, H.; Gyuris, T.; Chen, J.-L.; Tian, H.; Li, Y. Elucidation of signaling and functional activities of an orphan GPCR, GPR81. J. Lipid Res. 2008, 49, 797–803. [Google Scholar] [CrossRef]
- Sun, Z.; Han, Y.; Song, S.; Chen, T.; Han, Y.; Liu, Y. Activation of GPR81 by lactate inhibits oscillatory shear stress-induced endothelial inflammation by activating the expression of KLF2. IUBMB Life 2019, 71, 2010–2019. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Bergersen, L.H. Lactate transport and signaling in the brain: Potential therapeutic targets and roles in body-brain interaction. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Khatib-Massalha, E.; Bhattacharya, S.; Massalha, H.; Biram, A.; Golan, K.; Kollet, O.; Kumari, A.; Avemaria, F.; Petrovich-Kopitman, E.; Gur-Cohen, S.; et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat. Commun. 2020, 11, 3547. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Hata, K.; Hirose, K.; Okui, T.; Toyosawa, S.; Uzawa, N.; Nishimura, R.; Yoneda, T. The lactate sensor GPR81 regulates glycolysis and tumor growth of breast cancer. Sci. Rep. 2022, 12, 6261. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef]
- Nho, R.S.; Rice, C.; Prasad, J.; Bone, H.; Farkas, L.; Rojas, M.; Horowitz, J.C. Persistent hypoxia promotes myofibroblast differentiation via GPR-81 and differential regulation of LDH isoenzymes in normal and idiopathic pulmonary fibrosis fibroblasts. Physiol. Rep. 2023, 11, e15759. [Google Scholar] [CrossRef]
- Yang, L.; Gilbertsen, A.; Xia, H.; Benyumov, A.; Smith, K.; Herrera, J.; Racila, E.; Bitterman, P.B.; Henke, C.A. Hypoxia enhances IPF mesenchymal progenitor cell fibrogenicity via the lactate/GPR81/HIF1α pathway. JCI Insight 2023, 8, e163820. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Tang, C.; Müller, M.; Gille, A.; Sassmann, A.; Hanson, J.; Offermanns, S. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 2010, 11, 311–319. [Google Scholar] [CrossRef]
- Sunaga, H.; Matsui, H.; Ueno, M.; Maeno, T.; Iso, T.; Syamsunarno, M.R.A.A.; Anjo, S.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013, 4, 2563. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liang, C.; Liu, L.; Wang, L.; Yu, G. High-Fat Diet Related Lung Fibrosis-Epigenetic Regulation Matters. Biomolecules 2023, 13, 558. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.; Bellomo, R. A rational approach to fluid therapy in sepsis. Br. J. Anaesth. 2016, 116, 339–349. [Google Scholar] [CrossRef]
- Sladen, R.N. Lactate in sepsis and trauma—Hindrance or help? Anästhesiol. Intensivmed.·Notfallmed. Schmerzther. 1999, 34, 237–238. [Google Scholar] [CrossRef]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal 2007, 9, 1115–1124. [Google Scholar] [CrossRef]
- Ohno, Y.; Ando, K.; Ito, T.; Suda, Y.; Matsui, Y.; Oyama, A.; Kaneko, H.; Yokoyama, S.; Egawa, T.; Goto, K. Lactate Stimulates a Potential for Hypertrophy and Regeneration of Mouse Skeletal Muscle. Nutrients 2019, 11, 869. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Preclinical Data in IPF |
|---|---|---|
| PFKFB3 | 3PO | ↓ The activation of fibroblasts, |
| ↓ lung fibrosis in mice [66,90]. | ||
| HK2 | Lonidamine | ↓ TGF-β-induced fibrosis, |
| ↓ Fibrosis marker, | ||
| ↑ Lung function in mice [44]. | ||
| 2-DG | ↓ Lactate level, | |
| ↓ collagen synthesis [90]. | ||
| Glycolysis | KD025 | ↑ OXPHOS, |
| ↓ Lactate level, | ||
| ↓ pulmonary endothelial permeability [91]. | ||
| GLUT1 | GLUT inhibitor II | ↓ PAI-1, CTGF, and α-SMA [37]. |
| Phloretin | ↓ α-SMA, collagen, and fibronectin, | |
| ↓ lung fibrosis in mice [37,38]. | ||
| PDK1 | Dichloroacetate | ↓ Lactate and α-SMA production, |
| ↓ lung fibrosis in mice [92]. | ||
| PKM2 | Naphthoquinone derivatives | ↓ TGF-β signal, |
| ↓ collagen deposition [93]. | ||
| LDHA | Gossypol | Lactate production, |
| fibroblast differentiation, | ||
| pulmonary fibrosis in mice [94,95,96]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, P.; Liu, J.; Li, Z.; Wang, J.; Zhu, Z.; Wang, L.; Yu, G. Glycolysis Reprogramming in Idiopathic Pulmonary Fibrosis: Unveiling the Mystery of Lactate in the Lung. Int. J. Mol. Sci. 2024, 25, 315. https://doi.org/10.3390/ijms25010315
Yan P, Liu J, Li Z, Wang J, Zhu Z, Wang L, Yu G. Glycolysis Reprogramming in Idiopathic Pulmonary Fibrosis: Unveiling the Mystery of Lactate in the Lung. International Journal of Molecular Sciences. 2024; 25(1):315. https://doi.org/10.3390/ijms25010315
Chicago/Turabian StyleYan, Peishuo, Jingyi Liu, Zhenwei Li, Jiawei Wang, Zhao Zhu, Lan Wang, and Guoying Yu. 2024. "Glycolysis Reprogramming in Idiopathic Pulmonary Fibrosis: Unveiling the Mystery of Lactate in the Lung" International Journal of Molecular Sciences 25, no. 1: 315. https://doi.org/10.3390/ijms25010315
APA StyleYan, P., Liu, J., Li, Z., Wang, J., Zhu, Z., Wang, L., & Yu, G. (2024). Glycolysis Reprogramming in Idiopathic Pulmonary Fibrosis: Unveiling the Mystery of Lactate in the Lung. International Journal of Molecular Sciences, 25(1), 315. https://doi.org/10.3390/ijms25010315