
GABAA Receptor β3 Subunit Mutation N328D Heterozygous Knock-in Mice Have Lennox–Gastaut Syndrome
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
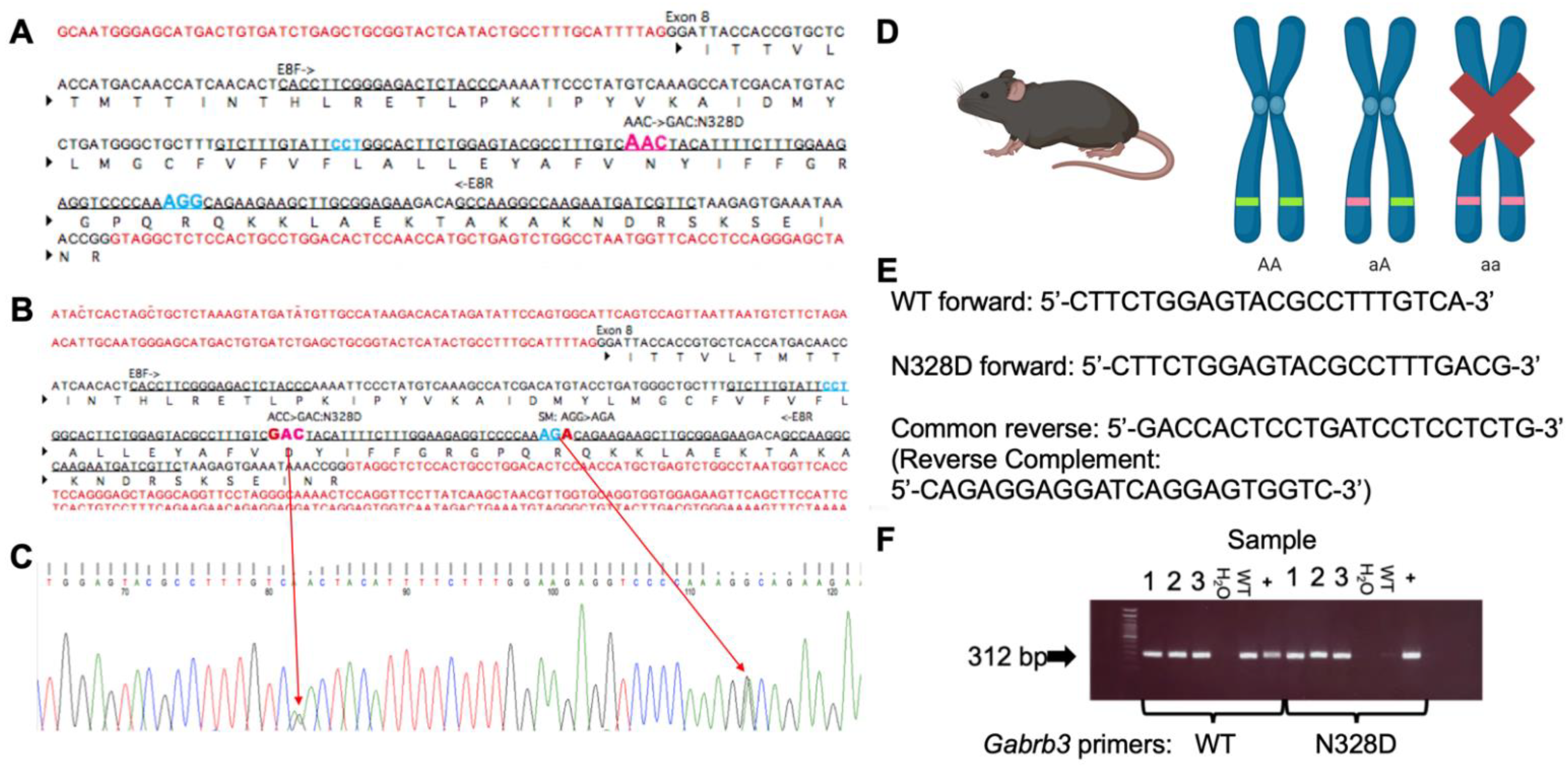
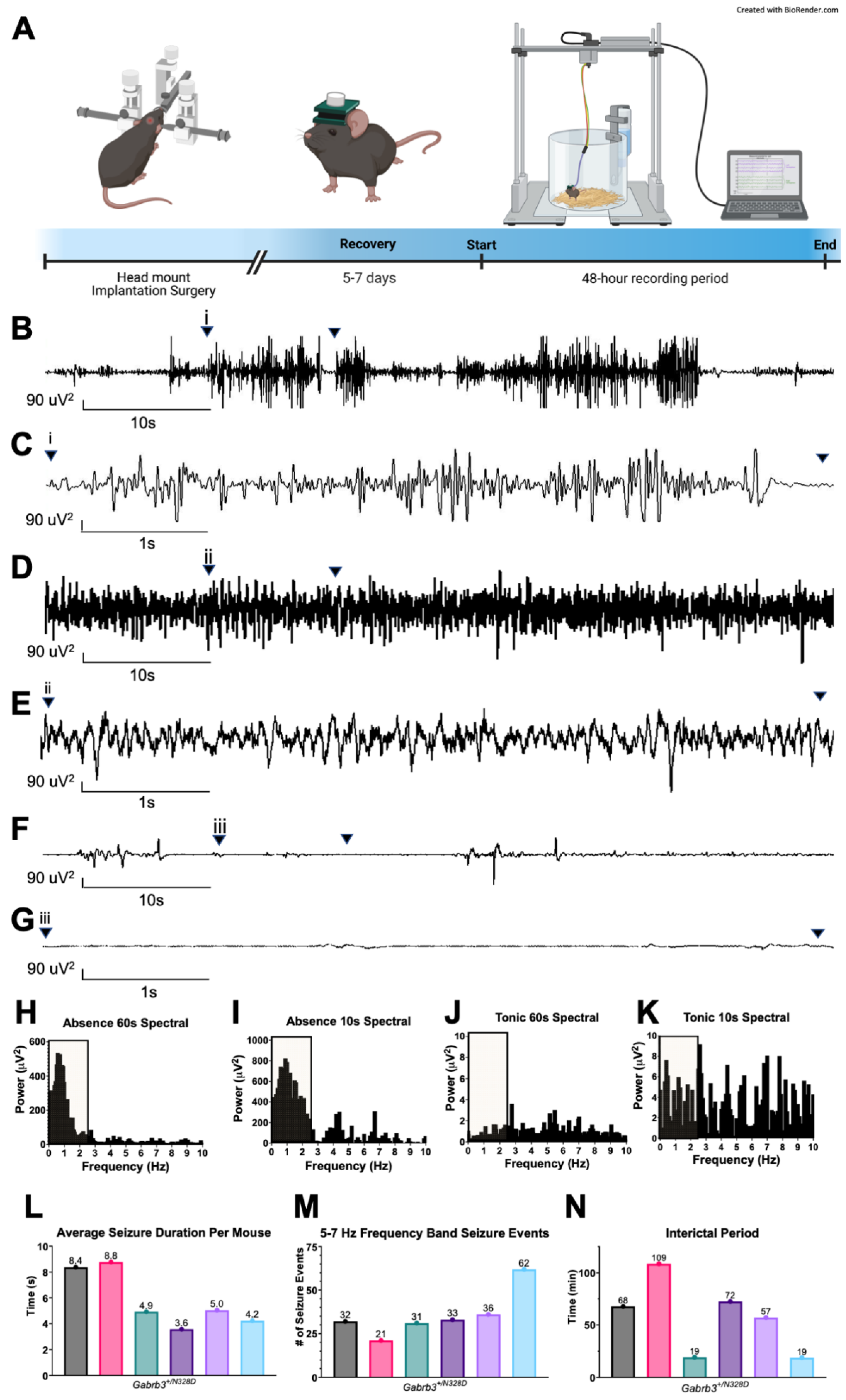
2.1. Gabrb3+/N328D Mice Displayed Seizure Types and Ictal EEG Patterns Consistent with LGS
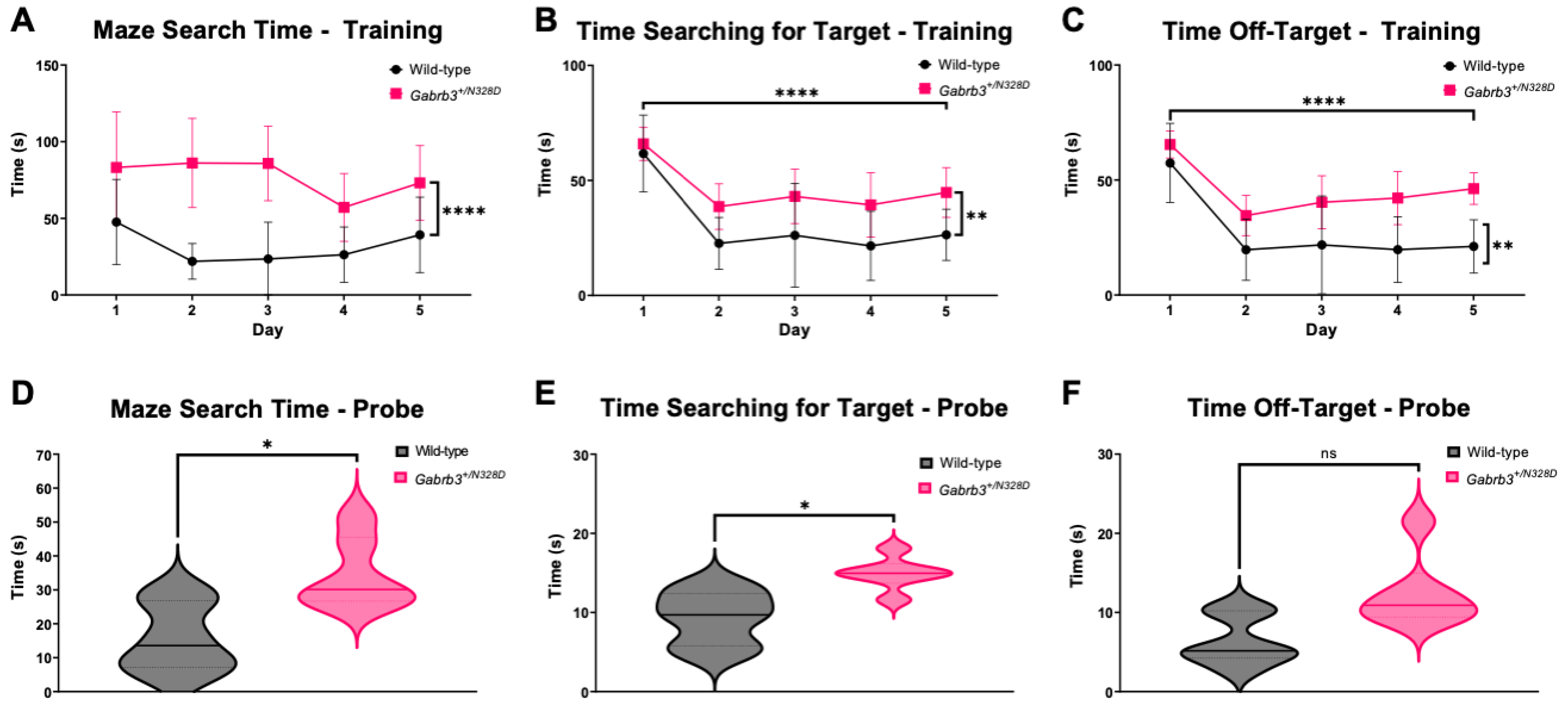
2.2. Gabrb3+/N328D Mice Had Impaired Learning Capabilities
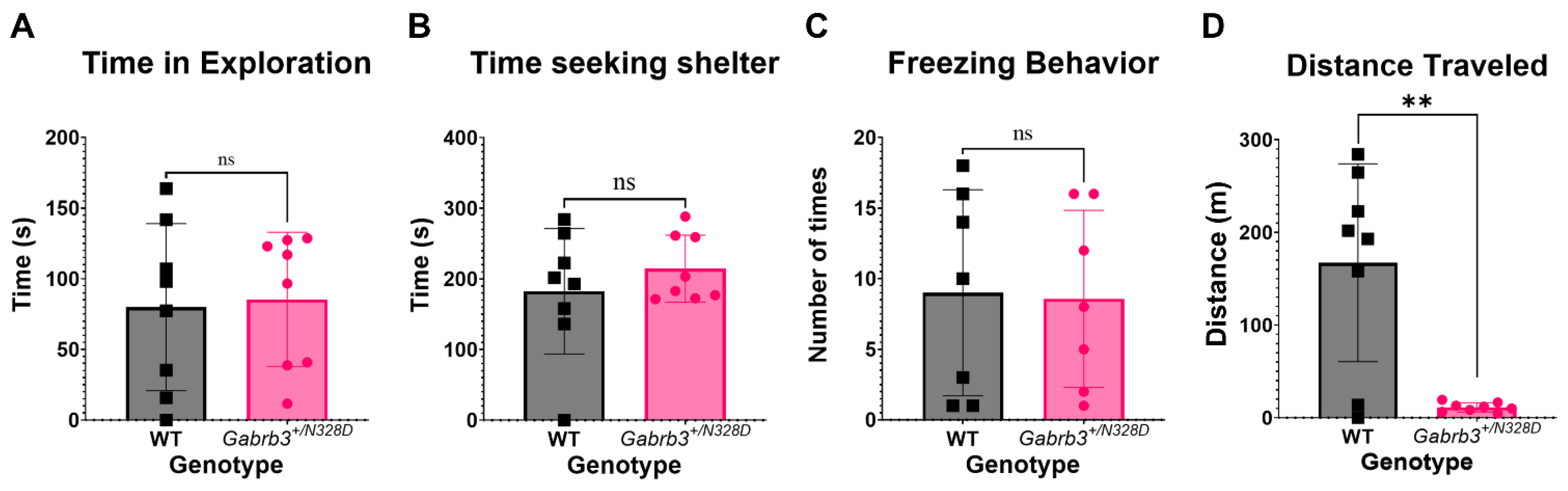
2.3. Gabrb3+/N328D Mice Did Not Display a Comorbid Autism Spectrum Disorder Phenotype
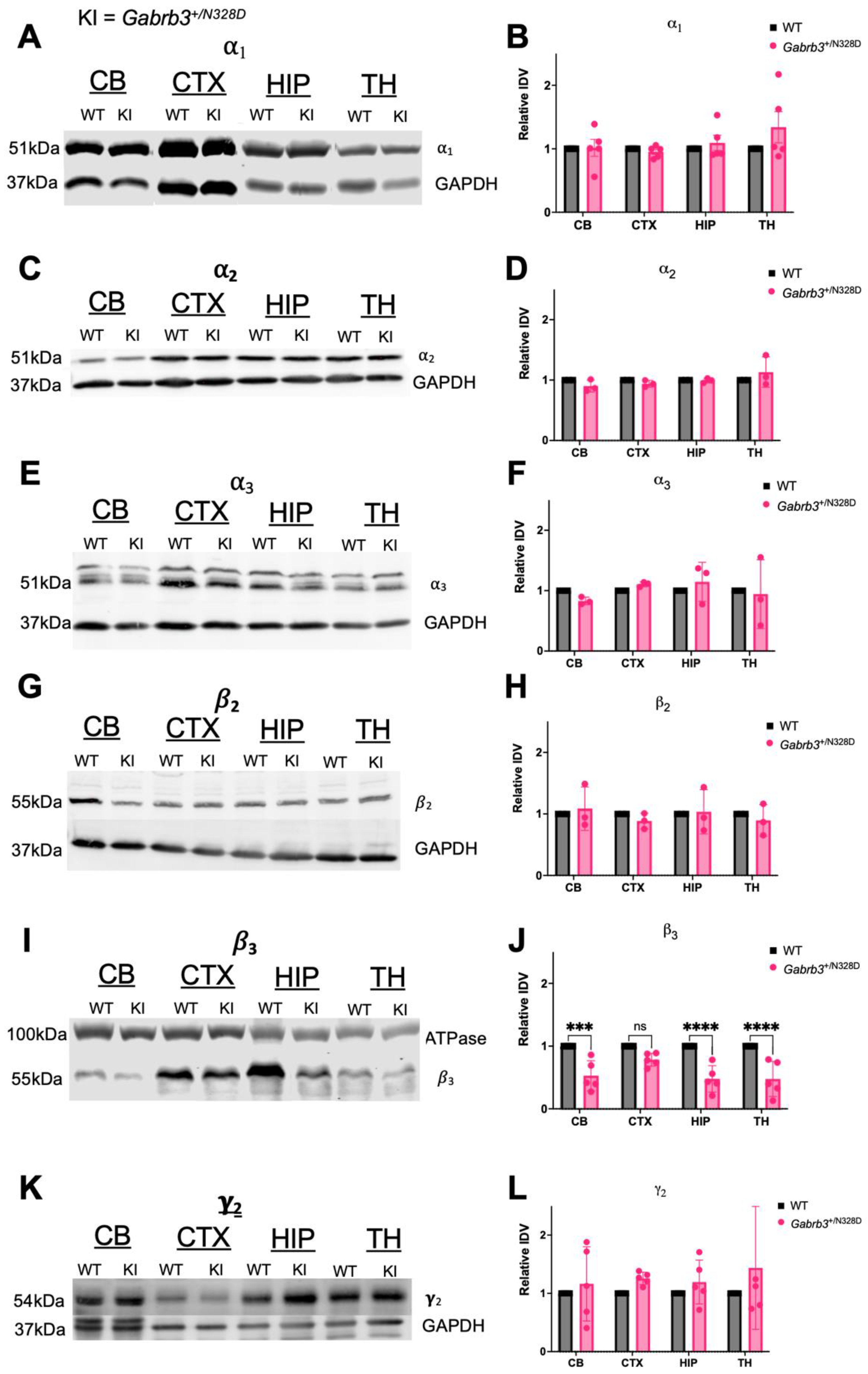
2.4. Gabrb3+/N328D Mice Showed Diminished Expression of β3 Subunit in Cerebellar, Thalamic, and Hippocampal Tissue
3. Discussion
3.1. Gabrb3+/N328D Mice Exhibit Seizure Types Typical of LGS
3.2. Gabrb3+/N328D Mice Showed a Cognitive Decline in Learning Ability and Deficit in Locomotion
3.3. Gabrb3+/N328D Mice Showed Reduced Expression of GABAAR β3 Subunit in Cerebellum, Hippocampus, and Thalamus
3.4. Gabrb3+/N328D Mice Are a Viable Model for Studying Lennox–Gastaut Syndrome Pathophysiology and Therapeutic Intervention
4. Materials and Methods
4.1. Subjects
4.2. Generation of the Gabrb3+/N328D Knock-in Mouse
4.3. Video-EEG Recordings and Analysis
4.4. Mouse Behavioral Testing
4.4.1. Three-Chamber Social Interaction Test
4.4.2. Barnes Maze
4.4.3. Elevated Zero Maze
4.4.4. Home Cage Monitoring
4.5. Western Blotting
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asadi-Pooya, A.A. Lennox-Gastaut syndrome: A comprehensive review. Neurol. Sci. 2017, 39, 403–414. [Google Scholar] [CrossRef] [PubMed]
- AOstendorf, A.P.; Ng, Y.-T. Treatment-resistant Lennox-Gastaut syndrome: Therapeutic trends, challenges and future directions. Neuropsychiatr. Dis. Treat. 2017, 13, 1131–1140. [Google Scholar] [CrossRef]
- He, N.; Li, B.-M.; Li, Z.-X.; Wang, J.; Liu, X.-R.; Meng, H.; Tang, B.; Bian, W.-J.; Shi, Y.-W.; Liao, W.-P. Few individuals with Lennox-Gastaut syndrome have autism spectrum disorder: A comparison with Dravet syndrome. J. Neurodev. Disord. 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Crumrine, P.K. Lennox-Gastaut Syndrome. J. Child Neurol. 2002, 17, S70–S75. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.R.; Trevathan, E.; Braun, K.V.N.; Yeargin-Allsopp, M. Increased Risk of Death among Children with Lennox-Gastaut Syndrome and Infantile Spasms. J. Child Neurol. 2009, 25, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Camfield, P.R. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M. Lennox–Gastaut Syndrome: A State-of-the-Art Review. Neuropediatrics 2017, 48, 143–151. [Google Scholar] [CrossRef]
- Nwosu, G.; Reddy, S.B.; Riordan, H.R.M.; Kang, J.-Q. Variable Expression of GABAA Receptor Subunit Gamma 2 Mutation in a Nuclear Family Displaying Developmental and Encephalopathic Phenotype. Int. J. Mol. Sci. 2022, 23, 9683. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Kang, J.-Q.; Gallagher, M.J. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 2010, 588, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-W.; Zhang, Q.; Cai, K.; Poliquin, S.; Shen, W.; Winters, N.; Yi, Y.-H.; Wang, J.; Hu, N.; Macdonald, R.L.; et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 2019, 142, 3028–3044. [Google Scholar] [CrossRef]
- Qu, S.; Catron, M.; Zhou, C.; Janve, V.; Shen, W.; Howe, R.K.; Macdonald, R.L. GABAA receptor β3 subunit mutation D120N causes Lennox–Gastaut syndrome in knock-in mice. Brain Commun. 2020, 2, fcaa028. [Google Scholar] [CrossRef]
- Homanics, G.E.; DeLorey, T.M.; Firestone, L.L.; Quinlan, J.J.; Handforth, A.; Harrison, N.L.; Krasowski, M.D.; Rick, C.E.M.; Korpi, E.R.; Mäkelä, R.; et al. Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc. Natl. Acad. Sci. USA 1997, 94, 4143–4148. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, A.; McTague, A.; Trump, N.; Ambegaonkar, G.; Ngoh, A.; Meyer, E.; Scott, R.H.; A Kurian, M. GABRB3 mutations: A new and emerging cause of early infantile epileptic encephalopathy. Dev. Med. Child Neurol. 2015, 58, 416–420. [Google Scholar] [CrossRef]
- Nelson, J.A.; Demarest, S.; Thomas, J.; Juarez-Colunga, E.; Knupp, K.G. Evolution of Infantile Spasms to Lennox-Gastaut Syndrome: What Is There to Know? J. Child Neurol. 2021, 36, 752–759. [Google Scholar] [CrossRef]
- Qu, S.; Jackson, L.G.; Zhou, C.; Shen, D.; Shen, W.; Nwosu, G.; Howe, R.; Catron, M.A.; Flamm, C.; Biven, M.; et al. Heterozygous GABA A receptor β3 subunit N110D knock-in mice have epileptic spasms. Epilepsia 2023. [Google Scholar] [CrossRef]
- Letts, V.A.; Beyer, B.J.; Frankel, W.N. Hidden in plain sight: Spike-wave discharges in mouse inbred strains. Genes Brain Behav. 2014, 13, 519–526. [Google Scholar] [CrossRef]
- Bare, M.A.; Glauser, T.A.; Strawsburg, R.H. Need for electroencephalogram video confirmation of atypical absence seizures in children with Lennox-Gastaut syndrome. J. Child Neurol. 1998, 13, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S. The New Classification of Seizures by the International League against Epilepsy 2017. Curr. Neurol. Neurosci. Rep. 2017, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Gastaut, H.; Zifkin, B.G. The risk of automobile accidents with seizures occurring while driving: Relation to seizure type. Neurology 1987, 37, 1613. [Google Scholar] [CrossRef]
- Schiff, N.; Labar, D.; Victor, J. Common dynamics in temporal lobe seizures and absence seizures. Neuroscience 1999, 91, 417–428. [Google Scholar] [CrossRef]
- Ren, L.-K.; Wu, L.-W.; Jin, L.-R.; Gao, W.; Shao, X.-Q. Characteristics of clinical manifestations and EEG of Lennox-Gastaut syndrome. Chin. J. Pediatr. 2003, 41, 7–10. [Google Scholar]
- Chan, K.F.; Jia, Z.; Murphy, P.A.; Burnham, W.M.; Cortez, M.A.; Snead, O.C. Learning and memory impairment in rats with chronic atypical absence seizures. Exp. Neurol. 2004, 190, 328–336. [Google Scholar] [CrossRef]
- Cortez, M.A.; McKerlie, C.; Snead, O.C. A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology 2001, 56, 341–349. [Google Scholar] [CrossRef]
- Pitkanen, A.; Buckmaster, P.; Galanopoulou, A.; Moshe, S. Models of Seizures and Epilepsy, 2nd ed.; Melanie Tucker, K.P., Ed.; Mara Conner: London, UK, 2006. [Google Scholar]
- Werhahn, K.J.; Noachtar, S.; Arnold, S.; Pfander, M.; Henkel, A.; Winkler, P.A.; Luders, H.O. Tonic seizures: Their significance for lateralization and frequency in different focal epileptic syndromes. Epilepsia 2000, 41, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.K.S.; Frey, L.C.; Britton, J.W.; Frey, L.C.; Hopp, J.L.; Korb, P.; Koubeissi, M.Z.; Lievens, W.E.; Pestana-Knight, E.M.; Louis, E.K.S. EEG in the Epilepsies; StatPearls Publishing LLC.: Florida, FL, USA, 2016. [Google Scholar]
- Gohma, H.; Kuramoto, T.; Matalon, R.; Surendran, S.; Tyring, S.; Kitada, K.; Sasa, M.; Serikawa, T. Absence-Like and Tonic Seizures in Aspartoacylase/Attractin Double-Mutant Mice. Exp. Anim. 2007, 56, 161–165. [Google Scholar] [CrossRef]
- Blume, W.T. Lennox-Gastaut syndrome: Potential mechanisms of cognitive regression. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Pillay, N.; Archer, J.S.; Badawy, R.A.; Flanagan, D.F.; Berkovic, S.F.; Jackson, G. Networks underlying paroxysmal fast activity and slow spike and wave in Lennox-Gastaut syndrome. Neurology 2013, 81, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Baumer, N.; Spence, S.J. Evaluation and Management of the Child with Autism Spectrum Disorder. Contin. Lifelong Learn. Neurol. 2018, 24, 248–275. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.K.; Grewal, S.S.; Fletcher, A.; Bill, D.J.; Dourish, C.T. Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology 1994, 116, 56–64. [Google Scholar] [CrossRef]
- Angelakos, C.C.; Tudor, J.C.; Ferri, S.L.; Jongens, T.A.; Abel, T. Home-cage hypoactivity in mouse genetic models of autism spectrum disorder. Neurobiol. Learn. Mem. 2019, 165, 107000. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.-M.; Brünig, I. Formation and plasticity of GABAergic synapses: Physiological mechanisms and pathophysiological implications. Pharmacol. Ther. 2003, 98, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-Receptor Trafficking and Synaptic Dysfunction in Brain Disorders. Front. Cell. Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Guzmán, B.C.-F.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L. Gamma-aminobutyric acid A receptors in Alzheimer’s disease: Highly localized remodeling of a complex and diverse signaling pathway. Neural Regen. Res. 2018, 13, 1362–1363. [Google Scholar] [CrossRef]
- Intusoma, U.; Abbott, D.F.; Masterton, R.A.J.; Stagnitti, M.R.; Newton, M.R.; Jackson, G.D.; Freeman, J.L.; Harvey, A.S.; Archer, J.S. Tonic seizures of Lennox-Gastaut syndrome: Periictal single-photon emission computed tomography suggests a corticopontine network. Epilepsia 2013, 54, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Maheshwari, A.; Noebels, J.; Smirnakis, S. Asynchronous suppression of visual cortex during absence seizures in stargazer mice. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef]
- Landau, W.M.; Kleffner, F.R. Syndrome of Acquired Aphasia with Convulsive Disorder in Children. Neurology 1957, 7, 523. [Google Scholar] [CrossRef]
- Novak, A.; Vizjak, K.; Rakusa, M. Cognitive Impairment in People with Epilepsy. J. Clin. Med. 2022, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Hingray, C.; McGonigal, A.; Kotwas, I.; Micoulaud-Franchi, J.-A. The Relationship between Epilepsy and Anxiety Disorders. Curr. Psychiatry Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- Oguni, H.; Hayashi, K.; Osawa, M. Long-Term Prognosis of Lennox-Gastaut Syndrome. Epilepsia 1996, 37, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Benke, D.; Fritschy, J.M.; Trzeciak, A.; Bannwarth, W.; Mohler, H. Distribution, prevalence, and drug binding profile of gamma-aminobutyric acid type A receptor subtypes differing in the beta-subunit variant. J. Biol. Chem. 1994, 269, 27100–27107. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.-M.; Mohler, H. GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995, 359, 154–194. [Google Scholar] [CrossRef]
- Ogris, W.; Lehner, R.; Fuchs, K.; Furtmüller, B.; Höger, H.; Homanics, G.E.; Sieghart, W. Investigation of the abundance and subunit composition of GABAA receptor subtypes in the cerebellum of alpha1-subunit-deficient mice. J. Neurochem. 2005, 96, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Janve, V.S.; Hernandez, C.C.; Verdier, K.M.; Hu, N.; Macdonald, R.L. Epileptic encephalopathy de novo GABRB mutations impair γ-aminobutyric acid type A receptor function. Ann. Neurol. 2016, 79, 806–825. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Kang, J.Q.; Gallagher, M.J. GABA A Receptor Subunit Mutations and Genetic Epilepsies; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012.
- McClelland, J.L.; McNaughton, B.L.; O’Reilly, R.C. Why there are complementary learning systems in the hippocampus and neocortex: Insights from the successes and failures of connectionist models of learning and memory. Psychol. Rev. 1995, 102, 419–457. [Google Scholar] [CrossRef]
- Hörtnagl, H.; Tasan, R.; Wieselthaler, A.; Kirchmair, E.; Sieghart, W.; Sperk, G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 2013, 236, 345–372. [Google Scholar] [CrossRef]
- Paz, J.T.; Huguenard, J. Microcircuits and their interactions in epilepsy: Is the focus out of focus? Nat. Neurosci. 2015, 18, 351–359. [Google Scholar] [CrossRef]
- Absalom, N.L.; Liao, V.W.Y.; Johannesen, K.M.H.; Gardella, E.; Jacobs, J.; Lesca, G.; Gokce-Samar, Z.; Arzimanoglou, A.; Zeidler, S.; Striano, P.; et al. Gain-of-function and loss-of-function GABRB3 variants lead to distinct clinical phenotypes in patients with developmental and epileptic encephalopathies. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- DeLorey, T.M.; Handforth, A.; Anagnostaras, S.G.; Homanics, G.E.; Minassian, B.A.; Asatourian, A.; Fanselow, M.S.; Delgado-Escueta, A.; Ellison, G.D.; Olsen, R.W. Mice Lacking the β3 Subunit of the GABAA Receptor Have the Epilepsy Phenotype and Many of the Behavioral Characteristics of Angelman Syndrome. J. Neurosci. 1998, 18, 8505–8514. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Gu, J.; Zhang, Y.; Wang, W.; Shen, H.; Chen, G.; Wang, X. Plic-1, a new target in repressing epileptic seizure by regulation of GABAAR function in patients and a rat model of epilepsy. Clin. Sci. 2015, 129, 1207–1223. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Shen, W.; Huang, X.; Liu, Z.; Macdonald, R.L.; Kang, J.-Q. Differential molecular and behavioural alterations in mouse models ofGABRG2haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum. Mol. Genet. 2016, 25, 3192–3207. [Google Scholar] [CrossRef]
- Mermer, F.; Poliquin, S.; Zhou, S.; Wang, X.; Ding, Y.; Yin, F.; Shen, W.; Wang, J.; Rigsby, K.; Xu, D.; et al. Astrocytic GABA transporter 1 deficit in novel SLC6A1 variants mediated epilepsy: Connected from protein destabilization to seizures in mice and humans. Neurobiol. Dis. 2022, 172, 105810. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.A.; Smith, N.K.; Kang, J.-Q. The therapeutic effect of stiripentol in Gabrg2+/Q390X mice associated with epileptic encephalopathy. Epilepsy Res. 2019, 154, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Lipina, T.; Vukobradovic, I.; Roder, J.; Woodgett, J.R. Assessment of social interaction behaviors. J. Vis. Exp. JoVE 2011, 48, e2473. [Google Scholar] [CrossRef]
- Harrison, F.E.; Reiserer, R.S.; Tomarken, A.J.; McDonald, M.P. Spatial and nonspatial escape strategies in the Barnes maze. Learn. Mem. Cold Spring Harb. N. Y. 2006, 13, 809–819. [Google Scholar] [CrossRef]
- Singh, K.; Bishnoi, M.; Kulkarni, S. Elevated zero maze: A paradigm to evaluate antianxiety effects of drugs. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 343–348. [Google Scholar] [CrossRef]
- ANY-maze. Available online: https://www.any-maze.com/ (accessed on 12 December 2022).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nwosu, G.I.; Shen, W.; Zavalin, K.; Poliquin, S.; Randhave, K.; Flamm, C.; Biven, M.; Langer, K.; Kang, J.-Q. GABAA Receptor β3 Subunit Mutation N328D Heterozygous Knock-in Mice Have Lennox–Gastaut Syndrome. Int. J. Mol. Sci. 2023, 24, 8458. https://doi.org/10.3390/ijms24098458
Nwosu GI, Shen W, Zavalin K, Poliquin S, Randhave K, Flamm C, Biven M, Langer K, Kang J-Q. GABAA Receptor β3 Subunit Mutation N328D Heterozygous Knock-in Mice Have Lennox–Gastaut Syndrome. International Journal of Molecular Sciences. 2023; 24(9):8458. https://doi.org/10.3390/ijms24098458
Chicago/Turabian StyleNwosu, Gerald Ikemefuna, Wangzhen Shen, Kirill Zavalin, Sarah Poliquin, Karishma Randhave, Carson Flamm, Marshall Biven, Katherine Langer, and Jing-Qiong Kang. 2023. "GABAA Receptor β3 Subunit Mutation N328D Heterozygous Knock-in Mice Have Lennox–Gastaut Syndrome" International Journal of Molecular Sciences 24, no. 9: 8458. https://doi.org/10.3390/ijms24098458
APA StyleNwosu, G. I., Shen, W., Zavalin, K., Poliquin, S., Randhave, K., Flamm, C., Biven, M., Langer, K., & Kang, J.-Q. (2023). GABAA Receptor β3 Subunit Mutation N328D Heterozygous Knock-in Mice Have Lennox–Gastaut Syndrome. International Journal of Molecular Sciences, 24(9), 8458. https://doi.org/10.3390/ijms24098458