
In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods

 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. In Silico Ligand-Based Approach: DRUDITONLINE
2.2. ADME Properties
2.3. In Silico Structure-Based Studies: Molecular Docking at the Catalytic Site of SARS-CoV-2 MPRO
2.4. Statistical Analysis: Principal Component Analysis (PCA)
2.5. In Silico Structure-Based Studies: Induced Fit Docking (IFD) into the Allosteric Site of SARS-CoV-2 MPRO
2.6. Molecular Dynamic Simulation
3. Materials and Methods
3.1. Ligand-Based Studies
Biotarget Predictor Tool (BPT)
3.2. Structure-Based Studies
3.2.1. Ligand Preparation
3.2.2. Protein Preparations
3.2.3. Docking Validation
3.2.4. Induced Fit Docking
3.2.5. Molecular Dynamic Simulation
3.3. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef] [PubMed]
- Kevadiya, B.D.; Machhi, J.; Herskovitz, J.; Oleynikov, M.D.; Blomberg, W.R.; Bajwa, N.; Soni, D.; Das, S.; Hasan, M.; Patel, M.; et al. Diagnostics for SARS-CoV-2 infections. Nat. Mater. 2021, 20, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Swain, B.; Verma, R.S.; Gunthe, S.S. SARS-CoV, MERS-CoV, and 2019-nCoV viruses: An overview of origin, evolution, and genetic variations. Virusdisease 2020, 31, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. InfezMed 2020, 28, 174–184. [Google Scholar]
- Bian, L.; Gao, F.; Zhang, J.; He, Q.; Mao, Q.; Xu, M.; Liang, Z. Effects of SARS-CoV-2 variants on vaccine efficacy and response strategies. Expert Rev. Vaccines 2021, 20, 365–373. [Google Scholar] [CrossRef]
- Drożdżal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Kotfis, K.; Ghavami, S.; Łos, M.J. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates 2020, 53, 100719. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Gorshkov, K.; Chen, C.Z.; Zheng, W. Drug Discovery Strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [Google Scholar] [CrossRef]
- Martorana, A.; Perricone, U.; Lauria, A. The Repurposing of Old Drugs or Unsuccessful Lead Compounds by in Silico Approaches: New Advances and Perspectives. Curr. Top. Med. Chem. 2016, 16, 2088–2106. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure-Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Cho, E.; Rosa, M.; Anjum, R.; Mehmood, S.; Soban, M.; Mujtaba, M.; Bux, K.; Moin, S.T.; Tanweer, M.; Dantu, S.; et al. Dynamic Profiling of β-Coronavirus 3CL Mpro Protease Ligand-Binding Sites. J. Chem. Inf. Model. 2021, 61, 3058–3073. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Fan, K.; Ma, L.; Han, X.; Liang, H.; Wei, P.; Liu, Y.; Lai, L. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2005, 329, 934–940. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef]
- Liang, J.; Karagiannis, C.; Pitsillou, E.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface. Comput. Biol. Chem. 2020, 89, 107372. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef]
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem. Int. Ed. 2020, 59, 23544–23548. [Google Scholar] [CrossRef]
- El Ahdab, D.; Lagardère, L.; Inizan, T.J.; Célerse, F.; Liu, C.; Adjoua, O.; Jolly, L.H.; Gresh, N.; Hobaika, Z.; Ren, P.; et al. Interfacial Water Many-Body Effects Drive Structural Dynamics and Allosteric Interactions in SARS-CoV-2 Main Protease Dimerization Interface. J. Phys. Chem. Lett. 2021, 12, 6218–6226. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, W.R.; Gomes, R.A.; Novaes, S.A.L.; Goulart Trossini, G.H. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: An in silico repurposing study. Future Med. Chem. 2020, 12, 1815–1828. [Google Scholar] [CrossRef]
- Somboon, T.; Mahalapbutr, P.; Sanachai, K.; Maitarad, P.; Lee, V.S.; Hannongbua, S.; Rungrotmongkol, T. Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J. Mol. Liq. 2021, 322, 114999. [Google Scholar] [CrossRef] [PubMed]
- Amendola, G.; Ettari, R.; Previti, S.; Di Chio, C.; Messere, A.; Di Maro, S.; Hammerschmidt, S.J.; Zimmer, C.; Zimmermann, R.A.; Schirmeister, T.; et al. Lead Discovery of SARS-CoV-2 Main Protease Inhibitors through Covalent Docking-Based Virtual Screening. J. Chem. Inf. Model. 2021, 61, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, N.; Chowdhury, P.; Goswami, A.K.; Das, A.; Chetia, D.; Gogoi, B. Computational guided identification of a citrus flavonoid as potential inhibitor of SARS-CoV-2 main protease. Mol. Divers. 2021, 25, 1745–1759. [Google Scholar] [CrossRef]
- Gupta, A.; Rani, C.; Pant, P.; Vijayan, V.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S.; Sharma, P. Structure-Based Virtual Screening and Biochemical Validation to Discover a Potential Inhibitor of the SARS-CoV-2 Main Protease. ACS Omega 2020, 5, 33151–33161. [Google Scholar] [CrossRef] [PubMed]
- Shivanika, C.; Kumar, S.D.; Ragunathan, V.; Tiwari, P.; Sumitha, A.; Devi, P.B. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2022, 40, 585–611. [Google Scholar] [CrossRef]
- Llanes, A.; Cruz, H.; Nguyen, V.D.; Larionov, O.V.; Fernández, P.L. A Computational Approach to Explore the Interaction of Semisynthetic Nitrogenous Heterocyclic Compounds with the SARS-CoV-2 Main Protease. Biomolecules 2020, 11, 18. [Google Scholar] [CrossRef]
- Sobhia, M.E.; Kumar, G.S.; Sivangula, S.; Ghosh, K.; Singh, H.; Haokip, T.; Gibson, J. Rapid structure-based identification of potential SARS-CoV-2 main protease inhibitors. Future Med. Chem. 2021, 13, 1435–1450. [Google Scholar] [CrossRef]
- Uniyal, A.; Mahapatra, M.K.; Tiwari, V.; Sandhir, R.; Kumar, R. Targeting SARS-CoV-2 main protease: Structure based virtual screening, in silico ADMET studies and molecular dynamics simulation for identification of potential inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 3609–3625. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Eid, E.E.M.; Almutairi, A. Targeting SARS-CoV-2 main protease by teicoplanin: A mechanistic insight by docking, MM/GBSA and molecular dynamics simulation. J. Mol. Struct. 2021, 1246, 131124. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S. New insights into the catalytic mechanism of the SARS-CoV-2 main protease: An ONIOM QM/MM approach. Mol. Divers. 2022, 26, 1373–1381. [Google Scholar] [CrossRef]
- Lodola, A.; Callegari, D.; Scalvini, L.; Rivara, S.; Mor, M. Design and SAR Analysis of Covalent Inhibitors Driven by Hybrid QM/MM Simulations. Methods Mol. Biol. 2020, 2114, 307–337. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.S.; Islam, R.; Parves, M.R.; Mamun, A.A.; Shahriar, I.; Hossain, M.I.; Hossain, M.N.; Ali, M.A.; Halim, M.A. Cysteine focused covalent inhibitors against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 1639–1658. [Google Scholar] [CrossRef]
- Xiong, M.; Nie, T.; Shao, Q.; Li, M.; Su, H.; Xu, Y. In silico screening-based discovery of novel covalent inhibitors of the SARS-CoV-2 3CL protease. Eur. J. Med. Chem. 2022, 231, 114130. [Google Scholar] [CrossRef]
- Joshi, T.; Pundir, H.; Sharma, P.; Mathpal, S.; Chandra, S. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2021, 39, 6728–6746. [Google Scholar] [CrossRef]
- Ahmad, S.; Usman Mirza, M.; Yean Kee, L.; Nazir, M.; Abdul Rahman, N.; Trant, J.F.; Abdullah, I. Fragment-based in silico design of SARS-CoV-2 main protease inhibitors. Chem. Biol. Drug Des. 2021, 98, 604–619. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; Wu, T.; Fang, M.; Niu, Z.; Wu, Z.; Xu, D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules 2022, 12, 746. [Google Scholar] [CrossRef]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Mannino, S.; Gentile, C.; Mannino, G.; Martorana, A.; Peri, D. DRUDIT: Web-based DRUgs DIscovery Tools to design small molecules as modulators of biological targets. Bioinformatics 2020, 36, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Gentile, C.; Perricone, U.; Piccionello, A.P.; Bartolotta, R.; Terenzi, A.; Pace, A.; Mingoia, F.; Almerico, A.M.; Lauria, A. Synthesis, antiproliferative activity, and in silico insights of new 3-benzoylamino-benzo[b]thiophene derivatives. Eur. J. Med. Chem. 2015, 90, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; Gentile, C.; Mingoia, F.; Palumbo Piccionello, A.; Bartolotta, R.; Delisi, R.; Buscemi, S.; Martorana, A. Design, synthesis, and biological evaluation of a new class of benzo[b]furan derivatives as antiproliferative agents, with in silico predicted antitubulin activity. Chem. Biol. Drug Des. 2018, 91, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Gentile, C.; Lauria, A. In Silico Insights into the SARS CoV-2 Main Protease Suggest NADH Endogenous Defences in the Control of the Pandemic Coronavirus Infection. Viruses 2020, 12, 805. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 13, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Schrödinger Release 2017-2, LigPrep; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Schrödinger, LLC. Schrödinger Release 2017-2, Schrödinger Suite 2017-2 Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB. Available online: www.rcsb.org (accessed on 29 June 2022).
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
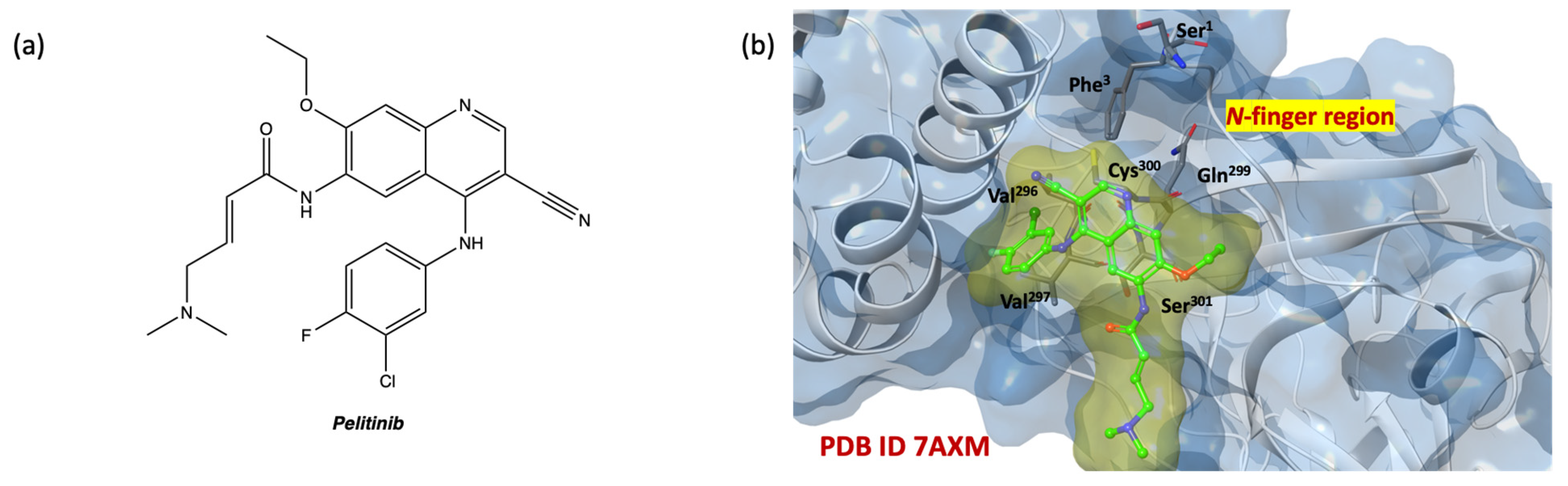{kind=link}
{kind=link}
| Compound | X | R1 | R2 | R3 | R4 | DAS Score |
|---|---|---|---|---|---|---|
| 2b | O | H | OCH3 | H | H | 0.940 |
| 2a | O | H | H | H | H | 0.922 |
| 2c | O | H | CH3 | H | H | 0.922 |
| 1b | S | H | OCH3 | H | H | 0.918 |
| 2g | O | H | H | H | CH3 | 0.916 |
| 2f | O | Cl | F | H | H | 0.910 |
| 2l | O | Cl | F | H | CH3 | 0.910 |
| 2i | O | H | CH3 | H | CH3 | 0.904 |
| 2h | O | H | OCH3 | H | CH3 | 0.900 |
| 1d | S | OCH3 | OCH3 | OCH3 | H | 0.900 |
| 1c | S | H | CH3 | H | H | 0.898 |
| 1f | S | Cl | F | H | H | 0.890 |
| 1g | S | H | H | H | CH3 | 0.888 |
| 1i | S | H | CH3 | H | CH3 | 0.882 |
| 1j | S | OCH3 | OCH3 | OCH3 | CH3 | 0.882 |
| 1l | S | Cl | F | H | CH3 | 0.880 |
| 2d | O | OCH3 | OCH3 | OCH3 | H | 0.880 |
| 1a | S | H | H | H | H | 0.880 |
| 1h | S | H | OCH3 | H | CH3 | 0.880 |
| 2k | O | H | CF3 | H | CH3 | 0.820 |
| 1e | S | H | CF3 | H | H | 0.820 |
| 2j | O | OCH3 | OCH3 | OCH3 | CH3 | 0.862 |
| 2e | O | H | CF3 | H | H | 0.836 |
| 1k | S | H | CF3 | H | CH3 | 0.800 |
| Compound | Lipinski Violations | Ghose Violations | Veber Violations | Egan Violations | PAINS Alerts | Total |
|---|---|---|---|---|---|---|
| 1a | 0 | 0 | 0 | 0 | 0 | 0 |
| 1b | 0 | 0 | 0 | 1 | 0 | 1 |
| 1c | 0 | 0 | 0 | 0 | 0 | 0 |
| 1d | 1 | 2 | 2 | 1 | 0 | 6 |
| 1e | 0 | 2 | 0 | 1 | 0 | 3 |
| 1f | 0 | 0 | 0 | 0 | 0 | 0 |
| 1g | 0 | 0 | 0 | 0 | 0 | 0 |
| 1h | 0 | 0 | 0 | 1 | 0 | 1 |
| 1i | 0 | 0 | 0 | 0 | 0 | 0 |
| 1j | 1 | 2 | 2 | 1 | 0 | 6 |
| 1k | 1 | 2 | 0 | 1 | 0 | 4 |
| 1l | 0 | 2 | 0 | 0 | 0 | 2 |
| 2a | 0 | 0 | 0 | 0 | 0 | 0 |
| 2b | 0 | 0 | 0 | 0 | 0 | 0 |
| 2c | 0 | 0 | 0 | 0 | 0 | 0 |
| 2d | 1 | 2 | 1 | 1 | 0 | 5 |
| 2e | 0 | 1 | 0 | 0 | 0 | 1 |
| 2f | 0 | 0 | 0 | 0 | 0 | 0 |
| 2g | 0 | 0 | 0 | 0 | 0 | 0 |
| 2h | 0 | 0 | 0 | 0 | 0 | 0 |
| 2i | 0 | 0 | 0 | 0 | 0 | 0 |
| 2j | 2 | 2 | 1 | 1 | 0 | 6 |
| 2k | 0 | 2 | 0 | 1 | 0 | 3 |
| 2l | 0 | 0 | 0 | 0 | 0 | 0 |
| SARS-CoV-2 MPRO (pdb Code 7VH8) | ||
|---|---|---|
| Title | IFD Score | Docking Score |
| 1d | −675.768 | −8.979 |
| 2l | −675.108 | −12.040 |
| 1j | −674.838 | −7.595 |
| 1f | −674.292 | −9.781 |
| 1i | −674.180 | −9.222 |
| 2i | −674.046 | −11.050 |
| 2h | −674.040 | −10.969 |
| 1l | −674.037 | −7.673 |
| 1a | −673.969 | −8.573 |
| 1k | −673.927 | −10.744 |
| 1c | −673.740 | −8.008 |
| 1b | −673.730 | −8.314 |
| nirmatrelvir | −673.142 | −10.169 |
| 2c | −673.071 | −10.733 |
| 1h | −673.014 | −7.862 |
| 2j | −672.880 | −9.567 |
| 2a | −672.879 | −10.861 |
| 1g | −672.752 | −8.145 |
| 1e | −672.547 | −8.150 |
| 2k | −672.538 | −10.328 |
| 2g | −672.284 | −10.226 |
| 2d | −672.184 | −9.634 |
| 2b | −671.756 | −10.415 |
| 2f | −671.736 | −10.352 |
| 2e | −671.460 | −9.900 |
| SARS-CoV-2 MPRO (pdb Code 7VH8) | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1′ | S1 | S2 | S3/S4 | ||||||||||||||||||||||
| Title | IFD Score | T25 | T26 | L27 | H41 | V42 | C145 | F140 | L141 | N142 | G143 | H163 | E166 | H172 | M49 | M165 | L167 | P168 | V186 | D187 | R188 | Q189 | T190 | Q192 | TOT |
| 1d | −675.768 | X | X | X | X | X | X | X | X | X | X | XX | X | X | X | X | 16 | ||||||||
| 2l | −675.108 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 19 | ||||
| 1j | −674.838 | X | X | X | X | X | X | X | XX | X | X | X | X | X | 14 | ||||||||||
| 1f | −674.292 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 16 | ||||||||
| 1i | −674.180 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | XX | X | X | 21 | |||
| 2i | −674.046 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 20 | |||
| 2h | −674.040 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 18 | |||||
| 1l | −674.037 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 18 | |||||
| 1a | −673.969 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 17 | ||||||
| 1k | −673.927 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 17 | ||||||
| 1c | −673.740 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 18 | |||||
| 1b | −673.730 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 17 | |||||||
| nirmatrelvir | −673.142 | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X | 15 | ||||||||
| Title | PC1 | PC2 | Distance |
|---|---|---|---|
| pelitinib | −0.62 | 4.23 | - |
| 1h | −1.70 | 3.92 | 1.15 |
| 2g | −0.98 | 2.56 | 1.76 |
| 2i | 0.70 | 2.91 | 1.90 |
| 2c | −0.67 | 1.76 | 2.53 |
| 1i | −2.98 | 5.49 | 2.66 |
| 1b | −3.08 | 2.73 | 2.92 |
| 1c | −4.63 | 4.16 | 4.02 |
| 2a | −2.85 | 0.91 | 4.05 |
| 2h | 2.46 | 1.59 | 4.09 |
| 1g | −4.78 | 4.90 | 4.22 |
| 1l | −3.35 | 0.69 | 4.53 |
| 2b | 0.87 | 0.01 | 4.53 |
| 2l | −0.06 | −1.55 | 5.86 |
| Title | IFD Score | Docking Score |
|---|---|---|
| 1c | −693.48 | −7.005 |
| 1b | −692.66 | −7.214 |
| 2l | −692.66 | −6.327 |
| 1l | −692.59 | −6.72 |
| 2i | −692.24 | −7.522 |
| 1g | −692.05 | −6.368 |
| 1i | −691.57 | −5.981 |
| 2b | −691.36 | −6.187 |
| pelitinib | −691.09 | −6.192 |
| 2c | −691.03 | −6.675 |
| 1h | −690.98 | −5.082 |
| 2g | −690.73 | −5.954 |
| 2h | −690.62 | −5.679 |
| 2a | −689.92 | −6.238 |
| SARS-CoV-2 MPRO (pdb Code 7AXM) | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | IFD Score | S1 | G2 | D153 | Y154 | T209 | A210 | I213 | N214 | I249 | P252 | L253 | A255 | Q256 | F294 | V296 | V297 | R298 | C300 | S301 | G302 | V303 | T304 | F305 | TOT |
| 1c | −693.48 | X | X | X | X | X | X | X | X | X | X | 10 | |||||||||||||
| 1b | −692.66 | X | X | X | X | X | X | X | X | XX | X | X | 12 | ||||||||||||
| 2l | −692.66 | X | X | X | X | X | X | X | XX | X | X | X | 12 | ||||||||||||
| 1l | −692.59 | X | X | X | X | X | X | X | X | X | X | X | 11 | ||||||||||||
| 2i | −692.24 | X | X | X | X | X | X | X | X | X | X | XX | X | X | X | 15 | |||||||||
| 1g | −692.05 | X | X | X | X | X | X | X | X | X | X | X | 11 | ||||||||||||
| 2h | −691.57 | X | X | X | X | X | X | X | X | 8 | |||||||||||||||
| 1i | −691.36 | X | X | X | X | X | X | X | X | X | X | X | 11 | ||||||||||||
| 2b | −691.09 | X | X | X | X | X | X | X | X | X | X | X | X | 12 | |||||||||||
| pelitinib | −693.48 | X | X | X | X | X | X | X | X | 8 | |||||||||||||||
| Radii Van der Waals Scaling | Side Chain Optimization | Energy Minimization | RMSD | ||||
|---|---|---|---|---|---|---|---|
| Receptor Van der Waals Scaling | Ligand Van der Waals Scaling | Partial Charge Cut-Off | Residue Refinement | Distance-Dependent Dielectric Constant | Maximum Number of Minimization Steps | pdb Code 7VH8 | pdb Code 7AXM |
| 1.50 | 1.50 | 0.75 | 3 Å | 0.5 | 20 | 0.87 Å | 0.86 Å |
| 1.25 | 1.25 | 0.50 | 3.5 Å | 0.75 | 40 | 0.73 Å | 0.75 Å |
| 1.00 | 1.00 | 0.35 | 4 Å | 1.00 | 60 | 0.66 Å | 0.68 Å |
| 0.75 | 0.75 | 0.25 | 4.5 Å | 1.50 | 80 | 0.59 Å | 0.57 Å |
| 0.50 | 0.50 | 0.15 | 5 Å | 2.0 | 100 | 0.51 Å | 0.51 Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, A.; Lauria, A.; La Monica, G.; Alamia, F.; Mingoia, F.; Martorana, A. In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods. Int. J. Mol. Sci. 2023, 24, 8377. https://doi.org/10.3390/ijms24098377
Bono A, Lauria A, La Monica G, Alamia F, Mingoia F, Martorana A. In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods. International Journal of Molecular Sciences. 2023; 24(9):8377. https://doi.org/10.3390/ijms24098377
Chicago/Turabian StyleBono, Alessia, Antonino Lauria, Gabriele La Monica, Federica Alamia, Francesco Mingoia, and Annamaria Martorana. 2023. "In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods" International Journal of Molecular Sciences 24, no. 9: 8377. https://doi.org/10.3390/ijms24098377
APA StyleBono, A., Lauria, A., La Monica, G., Alamia, F., Mingoia, F., & Martorana, A. (2023). In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods. International Journal of Molecular Sciences, 24(9), 8377. https://doi.org/10.3390/ijms24098377