
In Vivo and In Vitro Characterization of Close Analogs of Compound KA-11, a New Antiseizure Drug Candidate

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis of Target Compounds KA-232 and KA-228
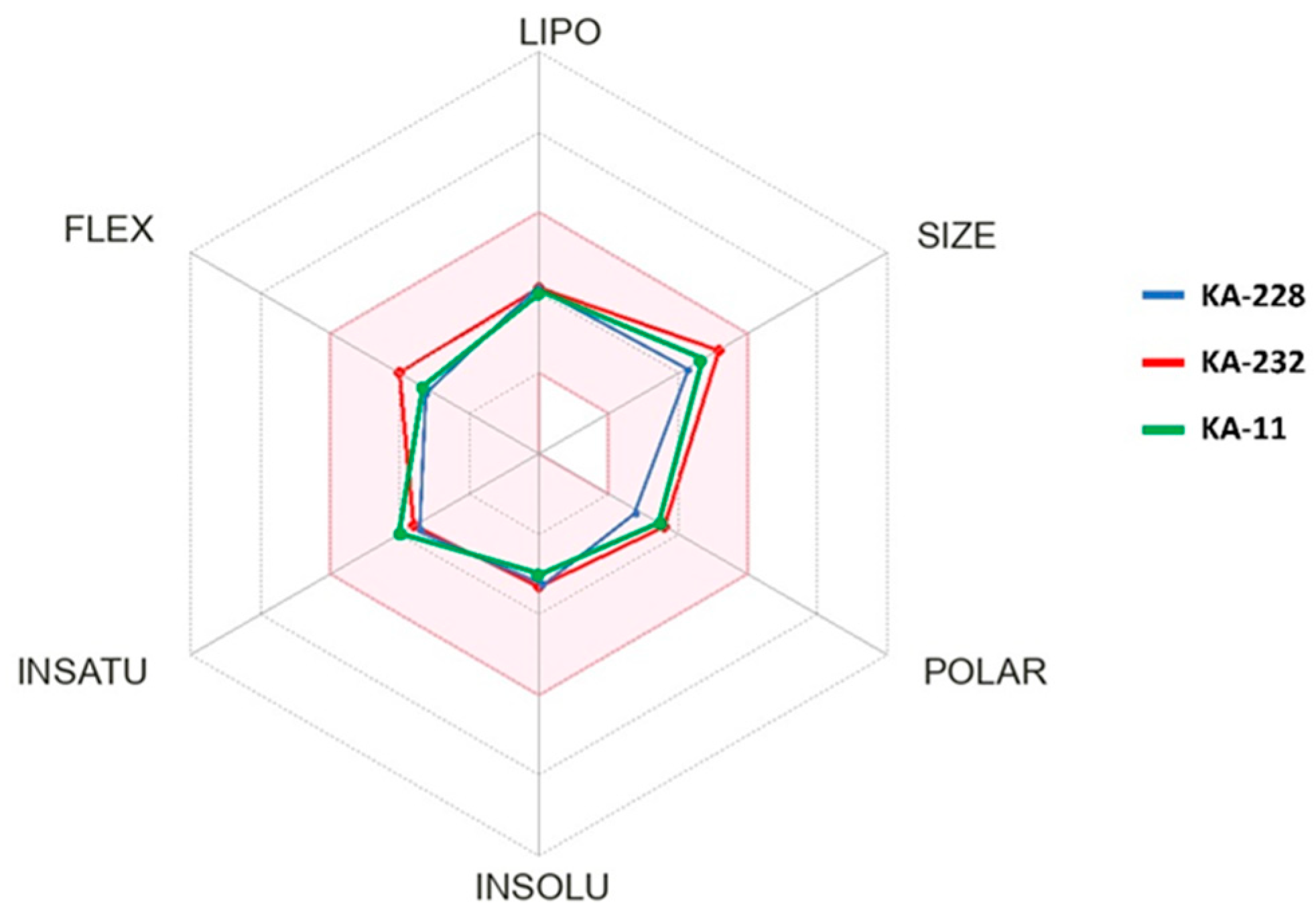
2.2. In Silico Determination of Selected Physicochemical Descriptors
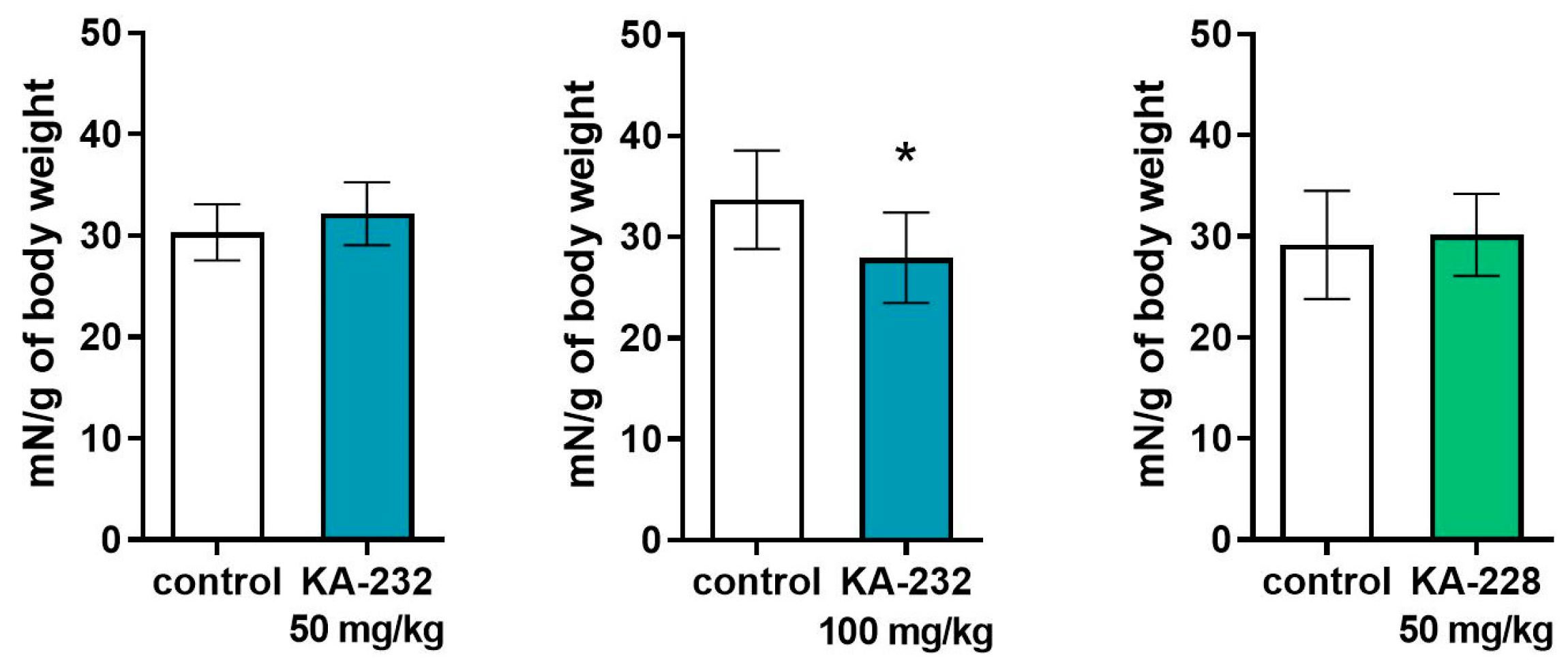
2.3. Antiseizure and Safety Evaluations In Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ED50 MES (mg/kg) ± SD | ED50 6 Hz (mg/kg) ± SD | ED50 PTZ (mg/kg) ± SD | TD50 (mg/kg) ± SD | TI |
|---|---|---|---|---|---|
| KA-11 [6] | 88.4 ± 34.16 | 21.0 ± 32.19 | 59.9 ± 19.55 | >1500 | >16.97 (MES) >71.43 (6 Hz) >25.04 (PTZ) |
| KA-228 | 92.5 ± 16.00 | 76.4 ± 40.76 **** | 95.4 ± 39.78 * | 131.40 ± 36.24 | 1.42 (MES) 1.72 (6 Hz) 1.38 (PTZ) |
| KA-232 | 61.8 ± 26.94 ** | 26.5 ± 33.46 | 59.3 ± 78.19 | 162.80 ± 31.20 | 2.63 (MES) 6.14 (6 Hz) 2.75 (PTZ) |
| VPA [8,19] | 355.2 ± 108.1 | 110.5 ± 42.8 | 176.2 ± 81.8 | 430.7 ± 47.1 | 1.21 (MES) 3.90 (6 Hz) 2.44 (PTZ) |
2.4. In Vitro Binding Assays
2.5. In Vitro Absorption Distribution Metabolism Excretion and Toxicity (ADME-Tox) Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthetic Procedure for Boc-Protected Compound (Tert-butyl-(1-oxo-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-2-yl)carbamate (1))
3.1.2. Synthetic Procedure for Amine (2-Amino-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-1-one (2))
3.1.3. Synthetic Procedure for Unsaturated Maleiamic Acid (4-Oxo-4-((1-oxo-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-2-yl)amino)but-2-enoic acid (3))
3.1.4. Synthetic Procedure for Unsaturated Pyrrolidine-2,5-Dione Derivative (1-(1-Oxo-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-2-yl)-1H-pyrrole-2,5-dione (4))
3.1.5. Synthetic Procedure for Target Water-Soluble Compound 3-(Dimethylamino)-1-(1-oxo-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-2-yl)pyrrolidine-2,5-dione hydrochloride (KA-232)
3.1.6. Synthetic Procedure for 2-((4-Bromobutyl)amino)-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-1-one (5)
3.1.7. Synthetic Procedure for Pyrrolidin-2-One Derivative 1-(1-Oxo-1-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propan-2-yl)pyrrolidin-2-one (KA-228)
3.2. In Silico Determination of Selected Physicochemical Descriptors
3.3. In Vivo Studies
3.3.1. Animals and Experimental Conditions
3.3.2. Drugs
3.3.3. Maximal Electroshock Seizure (MES) Test
3.3.4. 6-Hz (32 mA) Seizure Model
3.3.5. scPTZ-Induced Convulsions
3.3.6. Timed ivPTZ Seizure Threshold Test
3.3.7. Behavioral Tests
Chimney Test
Grip Strength Test
3.4. In Vitro Binding Assays
3.5. In Vitro ADME-Tox Studies
3.6. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | antiseizure drug |
| MES test | maximal electroshock seizure test |
| 6 Hz test | 6 Hertz test |
| scPTZ test | subcutaneous pentylenetetrazole test |
| ivPTZ test | intravenous pentylenetetrazole test |
| ADME-Tox | Absorption Distribution Metabolism Elimination and Toxicology |
| PILO | pilocarpine |
| PILO-induced SE | pilocarpine induced status epilepticus |
| DCC | (N,N′-dicyclohexylcarbodiimide) |
| TFA | trifluoroacetic acid |
| HMDS | hexamethyldisilazane |
| MW | molecular weight |
| Log P | lipholicity values |
| HBD | hydrogen bond donors |
| HBA | hydrogen bond acceptors |
| NBR | number of rotatable bonds |
| TPSA | topological polar surface area |
| LIPO | lipophilicity |
| POLAR | polarity |
| INSOLU | solubility |
| FLEX | flexibility |
| INSATU | saturation of the molecule |
| CNS | central nervous system |
| MPO | multiparameter optimization |
| GI | gastrointestinal |
| HIA | human intestinal absorption |
| BBB | blood-brain barrier |
| PGP+/− | poliglicoprotein |
| ED50 | effective dose |
| TD50 | Toxicity dose |
| SD | Standard Deviation |
| TI | Therapeutic Index |
| VPA | valproic acid |
| i.p. | intraperitoneal injection |
| GABAA | γ-aminobutyric acid A |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| NMDA | N-methyl-D-aspartic acid |
| hERG | human ether-à-go-go-related gene |
| TRPV1 | Transient receptor potential vanilloid 1 |
| VR1 | Vanilloid type 1 receptor |
| 5-HT1A | serotonin 1A |
| GAT-1 | GABA transporter 1 |
| SV2A | synaptic Vesicle Glycoprotein 2A |
| PAMPA | parallel artificial membrane permeability |
| HLMs | Human liver microsomes |
| CYP3A4 | Cytochrome P450 3A4 |
| CYP2D6 | Cytochrome P450 2D6 |
| HepG2 | Hepatoblastoma cell line |
| SH-SY5Y | Human Neuroblastoma cell line |
References
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, A.; Matricardi, S.; Mencaroni, E.; Dell’Isola, G.B.; Di Cara, G.; Striano, P.; Verrotti, A. The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front. Neurol. 2021, 12, 674483. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Guery, D.; Rheims, S. Is the mechanism of action of antiseizure drugs a key element in the choice of treatment? Fundam. Clin. Pharmacol. 2021, 35, 552–563. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Kamiński, K.; Zagaja, M.; Łuszczki, J.J.; Rapacz, A.; Andres-Mach, M.; Latacz, G.; Kieć- Kononowicz, K. Design, synthesis, and anticonvulsant activity of new hybrid compounds derived from 2-(2,5-dioxopyrrolidin-1-yl) propanamides and 2-(2,5 dioxopyrrolidin-1-yl) butanamides. J. Med. Chem. 2015, 58, 5274–5286. [Google Scholar] [CrossRef]
- Abram, M.; Jakubiec, M.; Reeb, K.; Cheng, M.H.; Gedschold, R.; Rapacz, A.; Mogilski, S.; Socała, K.; Nieoczym, D.; Szafarz, M.; et al. Discovery of (R)-N-Benzyl-2-(2,5-dioxopyrrolidin-1-yl)propanamide [(R)-AS-1], a Novel Orally Bioavailable EAAT2 Modulator with Drug-like Properties and Potent Antiseizure Activity In Vivo. J. Med. Chem. 2022, 65, 11703–11725. [Google Scholar] [CrossRef]
- Zagaja, M.; Szewczyk, A.; Szala-Rycaj, J.; Raszewski, G.; Chrościńska-Krawczyk, M.; Abram, M.; Kamiński, K.; Andres-Mach, M. C-11, a New Antiepileptic Drug Candidate: Evaluation of the Physicochemical Properties and Impact on the Protective Action of Selected Antiepileptic Drugs in the Mouse Maximal Electroshock-Induced Seizure Model. Molecules 2021, 26, 3144. [Google Scholar] [CrossRef]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a novel pyrrolidine- 2,5-dione derived broad-spectrum anticonvulsant: Its antiepileptogenic, antinociceptive properties and in vitro characterization. ACS Chem. Neurosci. 2019, 10, 636–648. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Luszczki, J.; Maj, M.; Rola, R.; Abram, M.; Kaminski, K. Evaluation of the impact of compound C11 a new anticonvulsant candidate on cognitive functions and hippocampal neurogenesis in mouse brain. Neuropharmacology 2020, 163, 107849. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Szala-Rycaj, J.; Lemieszek, M.; Maj, M.; Abram, M.; Kaminski, K. Preclinical assessment of a new hybrid compound C11 efficacy on neurogenesis and cognitive functions after pilocarpine induced status epilepticus in mice. Int. J. Mol. Sci. 2021, 22, 3240. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Swiss ADME Website. Available online: http://www.swissadme.ch/ (accessed on 10 January 2021).
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef]
- Löscher, W. Single-Target Versus Multi-Target Drugs Versus Combinations of Drugs with Multiple Targets: Preclinical and Clinical Evidence for the Treatment or Prevention of Epilepsy. Front. Pharmacol. 2021, 12, 730257. [Google Scholar] [CrossRef]
- Castel-Branco, M.M.; Alves, G.L.; Figueiredo, I.V.; Falcão, A.C.; Caramona, M.M. The maximal electroshock seizure (MES) model in the preclinical assessment of potential new antiepileptic drugs. Methods Find Exp. Clin. Pharmacol. 2009, 31, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Zagaja, M.; Zagaja, A.; Szala-Rycaj, J.; Szewczyk, A.; Lemieszek, M.K.; Raszewski, G.; Andres-Mach, M. Influence of Umbelliferone on the Anticonvulsant and Neuroprotective Activity of Selected Antiepileptic Drugs: An In Vivo and In Vitro Study. Int. J. Mol. Sci. 2022, 23, 3492. [Google Scholar] [CrossRef]
- Kamiński, K.; Socała, K.; Zagaja, M.; Andres-Mach, M.; Abram, M.; Jakubiec, M.; Pieróg, M.; Nieoczym, D.; Rapacz, A.; Gawel, K.; et al. N-Benzyl-(2,5-dioxopyrrolidin-1-yl)propanamide (AS-1) with Hybrid Structure as a Candidate for a Broad-Spectrum Antiepileptic Drug. Neurotherapeutics 2020, 17, 309–328. [Google Scholar] [CrossRef]
- Van Erum, J.; Van Dam, D.; De Deyn, P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019, 95, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Nieoczym, D.; Kowalczuk-Vasilev, E.; Wyska, E.; Wlaź, P. Increased seizure susceptibility and other toxicity symptoms following acute sulforaphane treatment in mice. Toxicol. Appl. Pharmacol. 2017, 326, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Nicita, F.; Spalice, A.; Raucci, U.; Iannetti, P.; Parisi, P. The possible use of the L-type calcium channel antagonist verapamil in drug-resistant epilepsy. Expert Rev. Neurother. 2016, 16, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, D.; Jha, S.K.; Jha, N.K.; Ambasta, R.K. Ion Channels in Neurological Disorders. Adv. Protein Chem. Struct. Biol. 2016, 103, 97–136. [Google Scholar]
- Löscher, W.; Klein, P. The Pharmacology and Clinical Efficacy of Antiseizure Medications: From Bromide Salts to Cenobamate and Beyond. CNS Drugs 2021, 35, 935–963. [Google Scholar] [CrossRef] [PubMed]
- Góra, M.; Czopek, A.; Rapacz, A.; Giza, A.; Koczurkiewicz-Adamczyk, P.; Pękala, E.; Obniska, J.; Kamiński, K. Design, Synthesis and Biological Activity of New Amides Derived from 3-Benzhydryl and 3-sec-Butyl-2,5-dioxo-pyrrolidin-1-yl-acetic Acid. ChemMedChem 2021, 16, 1619–1630. [Google Scholar] [CrossRef]
- Góra, M.; Czopek, A.; Rapacz, A.; Dziubina, A.; Głuch-Lutwin, M.; Mordyl, B.; Obniska, J. Synthesis, Anticonvulsant and Antinociceptive Activity of New Hybrid Compounds: Derivatives of 3-(3-Methylthiophen-2-yl)-pyrrolidine-2,5-dione. Int. J. Mol. Sci. 2020, 21, 5750. [Google Scholar] [CrossRef]
- Abram, M.; Rapacz, A.; Latacz, G.; Szulczyk, B.; Kalinowska-Tłuścik, J.; Otto-Ślusarczyk, D.; Struga, M.; Kamiński, R.M.; Kamiński, K. Asymmetric synthesis and in vivo/in vitro characterization of new hybrid anticonvulsants derived from (2,5-dioxopyrrolidin-1-yl)phenylacetamides. Bioorg. Chem. 2021, 109, 104751. [Google Scholar] [CrossRef]
- Abram, M.; Zagaja, M.; Mogilski, S.; Andres-Mach, M.; Latacz, G.; Baś, S.; Łuszczki, J.J.; Kieć-Kononowicz, K.; Kamiński, K. Multifunctional Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-yl)-3-methoxypropanamides with Anticonvulsant and Antinociceptive Properties. J. Med. Chem. 2017, 60, 8565–8579. [Google Scholar] [CrossRef]
- Abram, M.; Jakubiec, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Szulczyk, B.; Szafarz, M.; Socała, K.; Nieoczym, D.; Wyska, E.; et al. Identification of New Compounds with Anticonvulsant and Antinociceptive Properties in a Group of 3-substituted (2,5-dioxo-pyrrolidin-1-yl)(phenyl)-Acetamides. Int. J. Mol. Sci. 2021, 22, 13092. [Google Scholar] [CrossRef]
- Yang, F.; Sivils, A.; Cegielski, V.; Singh, S.; Chu, X.P. Transient Receptor Potential (TRP) Channels in Pain, Neuropsychiatric Disorders, and Epilepsy. Int. J. Mol. Sci. 2023, 24, 4714. [Google Scholar] [CrossRef]
- Fitzi-Rathgen, J. The 3Rs and replacement methods—Better research, less animal harm. ALTEX 2019, 36, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Zagaja, M.; Andres-Mach, M.; Patrzylas, P.; Pyrka, D.; Szpringer, M.; Florek-Łuszczki, M.; Żółkowska, D.; Skalicka-Woźniak, K.; Łuszczki, J.J. Influence of xanthotoxin (8-methoxypsoralen) on the anticonvulsant activity of various novel antiepileptic drugs against maximal electroshock-induced seizures in mice. Fitoterapia 2016, 115, 86–91. [Google Scholar] [CrossRef]
- Zagaja, M.; Andres-Mach, M.; Skalicka-Woźniak, K.; Rękas, A.R.; Kondrat-Wróbel, M.W.; Gleńsk, M.; Łuszczki, J.J. Assessment of the Combined Treatment with Umbelliferone and Four Classical Antiepileptic Drugs Against Maximal Electroshock-Induced Seizures in Mice. Pharmacology 2015, 96, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zagaja, M.; Bryda, J.; Szewczyk, A.; Szala-Rycaj, J.; Łuszczki, J.J.; Walczak, M.; Kuś, K.; Andres-Mach, M. Xanthotoxin enhances the anticonvulsant potency of levetiracetam and valproate in the 6-Hz corneal stimulation model in mice. Fundam. Clin. Pharmacol. 2022, 36, 133–142. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Zolkowska, D.; Barcicka-Klosowska, B.; Haratym-Maj, A.; Florek-Luszczki, M.; Luszczki, J.J. Effect of ACEA—A selective cannabinoid CB1 receptor agonist on the protective action of different antiepileptic drugs in the mouse pentylenetetrazole-induced seizure model. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 301–309. [Google Scholar] [CrossRef]
- Boissier, J.R.; Tardy, J.; Diverres, J.C. Une nouvelle methode simple pour explorer l’action tranquilisante: Le test de la cheminee. Med. Exp. 1960, 3, 81–84. [Google Scholar]
- Brown, G.B. 3H-batrachotoxinin-A benzoate binding to voltage-sensitive sodium channels: Inhibition by the channel blockers tetrodotoxin and saxitoxin. J. Neurosci. 1986, 6, 2064–2070. [Google Scholar] [CrossRef]
- Gould, R.J.; Murphy, K.M.; Snyder, S.H. [3H]nitrendipine-labeled calcium channels discriminate inorganic calcium agonists and antagonists. Proc. Natl. Acad. Sci. USA 1982, 79, 3656–3660. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, H.; Langer, S.Z. [3H]diltiazem binding to calcium channel antagonists recognition sites in rat cerebral cortex. Eur. J. Pharmacol. 1985, 111, 273–277. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Snowman, A.M.; Snyder, S.H. (-)-[3H] desmethoxyverapamil labels multiple calcium channel modulator receptors in brain and skeletal muscle membranes: Differentiation by temperature and dihydropyridines. J. Pharmacol. Exp. Ther. 1986, 237, 731–738. [Google Scholar]
- Sills, M.A.; Fagg, G.; Pozza, M.; Angst, C.; Brundish, D.E.; Hurt, S.D.; Wilusz, E.J.; Williams, M. [3H]CGP 39653: A new N-methyl-D-aspartate antagonist radioligand with low nanomolar affinity in rat brain. Eur. J. Pharmacol. 1991, 192, 19–24. [Google Scholar] [CrossRef]
- Murphy, D.E.; Snowhill, E.W.; Williams, M. Characterization of quisqualate recognition sites in rat brain tissue using DL-[3H]alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and a filtration assay. Neurochem. Res. 1987, 12, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Snowman, A.M.; Biswas, A.; Olivera, B.M.; Snyder, S.H. Omega-conotoxin GVIA binding to a high-affinity receptor in brain: Characterization, calcium sensitivity, and solubilization. J. Neurosci. 1988, 8, 3354–3359. [Google Scholar] [CrossRef]
- Wang, X.K. Pharmacological study on recombinant human GABA-A receptor complex containing alpha5 (leucine155 to valine) combined with beta3gamma2s subunits. Acta Pharmacol. Sin. 2001, 22, 521–523. [Google Scholar] [PubMed]
- Available online: https://www.eurofinsdiscovery.com/catalogmanagement/viewItem/271110 (accessed on 10 January 2021).
- Huang, X.P.; Mangano, T.; Hufeisen, S.; Setola, V.; Roth, B.L. Identification of human Ether-à-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 2010, 8, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Phelps, P.T.; Anthes, J.C.; Correll, C.C. Cloning and functional characterization of dog transient receptor potential vanilloid receptor-1 (TRPV1). Eur. J. Pharmacol. 2005, 513, 57–66. [Google Scholar] [CrossRef]
- Litchfield, J.T., Jr.; Wilcoxon, F. A simplified method of evaluating dose–effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
- Johannessen Landmark, C. Antiepileptic drugs in non-epilepsy disorders: Relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. [Google Scholar] [CrossRef]
- Johannessen Landmark, C.; Larsson, P.G.; Rytter, E.; Johannessen, S.I. Antiepileptic drugs in epilepsy and other disorders—A population-based study of prescriptions. Epilepsy Res. 2009, 87, 31–39. [Google Scholar] [CrossRef]
- Baftiu, A.; Johannessen Landmark, C.; Rusten, I.R.; Feet, S.A.; Johannessen, S.I.; Larsson, P.G. Changes in utilisation of antiepileptic drugs in epilepsy and non-epilepsy disorders—A pharmacoepidemiological study and clinical implications. Eur. J. Clin. Pharmacol. 2016, 72, 1245–1254. [Google Scholar] [CrossRef] [PubMed]










| Compound | Lipinski Rule | Veber Rule | CNS MPO | ||||
|---|---|---|---|---|---|---|---|
| MW a ≤500 | Log P b ≤5 | HBD c ≤5 | HBA d ≤10 | NBR e ≤10 | TPSA f ≤140 Å2 | ||
| KA-232 | 426.43 | 1.82 | 0 | 7 | 6 | 64.17 | 5.53 |
| KA-228 | 369.38 | 2.41 | 0 | 5 | 5 | 43.86 | 5.93 |
| KA-11 [8] | 383.37 | 1.98 | 0 | 6 | 5 | 60.93 | 5.83 |
| Binding Studies | Source | % Inhibition of Control Specific Binding (Concentration (µM)) a | ||
|---|---|---|---|---|
| KA-228 | KA-232 | KA-11 | ||
| Na+ channel (site 2) | Rat cerebral cortex | 15.5 (100) −2.9 (10) | 26.2 (100) 3.8 (10) | 37.4 (100) * |
| Calcium Cav1.2 channels (dihydropyridine site, antagonist radioligand) | Rat cerebral cortex | 32.5 (100) 8.7 (10) | 18.5 (100) −6.3 (10) | 64.2 (100) * |
| Calcium Cav1.2 channels (diltiazem site, antagonist radioligand) | Rat cerebral cortex | NT | 17.5 (100) −1.8 (10) | 44.9 (100) # |
| Calcium Cav1.2 channel (verapamil site, antagonist radioligand) | Rat cerebral cortex | NT | −2.8 (10) | 1.5 (100) # |
| Glutamate (NMDA, nonselective, antagonist radioligand, [3H] Ifenprodil radioligand) | Rat cerebral cortex | NT | 15.4 (10) | 13.0 (100) # |
| Glutamate (AMPA, nonselective, antagonist radioligand, [3H] AMPA radioligand) | Rat cerebral cortex | NT | 3.7 (10) | −2.9 (100) # |
| Calcium Cav2.2 channel (antagonist radioligand) | Rat cerebral cortex | NT | 10.6 (10) | 13.9 (100) * |
| GABAA (α1,β2,γ2) (agonist radioligand) | Human recombinant receptor | NT | −6.7 (10) | −28.1 (100) # |
| 5-HT1A (agonist radioligand) | Human recombinant receptor | NT | 9.6 (10) | 19.5 (100) # |
| Potassium channel (hERG) | Human recombinant HEK-293 cells | NT | 18.3 (100) | 15.0 (100) ** |
| Functional studies | Source | % Inhibition of control specific binding (concentration (µM)) a | ||
| KA-228 | KA-232 | KA-11 | ||
| TRPV1 (VR1) (antagonist effect) | Human recombinant CHO cells. | NT | 10.5 (10) | 33.0 (100) ** |
| ADME-Tox Assay In Vitro | KA-232 | KA-11 |
|---|---|---|
| PAMPA Pe (10−6 cm/s] ± SD | 10.20 ± 1.65 a | 27.1 ± 3.8 b [9] |
| Metabolic stability (% remaining after incubation with HLMs) | 73.81 | 73.49 [6] |
| CYP3A4 activity (% of control ± SD at 10 μM) | 86.77 ± 5.71 | 99.21 ± 4.9 [6] |
| CYP2D6 activity (% of control ± SD at 10 μM) | 100.96 ± 2.57 | 122.30 ± 2.02 [9] |
| HepG2 viability (% of control ± SD at 10 μM) | 89.44 ± 10.22 | 99.78 ± 2.18 [9] |
| SH-SY5Y viability (% of control ± SD at 10 μM) | 99.44 ± 5.89 | not tested |
| Assay/Ion Channels | Source | Ligand | Conc. (nM) | Kd (nM) | Non-Specific | Incubation | Detection Method | Ref. |
|---|---|---|---|---|---|---|---|---|
| Na+ channel (site 2) (antagonist radioligand) | rat cerebral cortex | [3H]batracho- toxin | 10 | 91 | veratridine (300 µM) | 60 min 37 °C | Scintillation counting | [38] |
| Calcium Cav1.2 channel (dihydropyridine site) (antagonist radioligand) | rat cerebral cortex | [3H]nitrendi- pine | 0.25 | 0.27 | nitrendipine (1 µM) | 90 min RT | Scintillation counting | [39] |
| Calcium Cav1.2 channel (diltiazem site) (benzothiazepines) (antagonist radioligand) | rat cerebral cortex | [3H]diltiazem | 15 | 52 | diltiazem (10 µM) | 120 min RT | Scintillation counting | [40] |
| Calcium Cav1.2 channel (verapamil site, antagonist radioligand) | rat cerebral cortex | [3H](-)- desmethoxy- verapamil | 0.4 | 14 | methoxyvera- pamil (10 µM) | 60 min RT | Scintillation counting | [41] |
| Glutamate (NMDA, nonselective, antagonist radioligand | rat cerebral cortex | [3H]ifenprodil | 2 | 26 | ifenprodil (10 µM) | 120 min 4 °C | Scintillation counting | [42] |
| Glutamate (AMPA, nonselective, antagonist radioligand) | rat cerebral cortex | [3H]AMPA | 5 | 18 | L-glutamic acid (1000 µM) | 90 min 4 °C | Scintillation counting | [43] |
| Calcium Cav2.2 channel (antagonist radioligand) | rat cerebral cortex | [125I]ω-conotoxin GVIA | 0.01 | 0.051 | w-conotoxin GVIA (0.1 µM) | 30 min 4 °C | Scintillation counting | [44] |
| GABAA (α1,β2,γ2) (agonist radioligand) | human recombinant receptor | [3H]muscimol | 15 | 30 | muscimol (10 µM) | 120 min RT | Scintillation counting | [45] |
| 5-HT1A (agonist radioligand) | human recombinant receptor | [3H]8-OH-DPAT | 1.5 | 2 | metergoline (10 µM) | 60 min RT | Scintillation counting | [46] |
| Potassium channel (hERG) | human recombinant HEK-293 cells | [3H]dofetilide | 3 | 6.6 | dofetilide (10 µM) | 60 min RT | Scintillation counting | [47] |
| Assay/Ion Channel | Source | Control Activator | Control Inhibitor | Measured Component | Incubation | Detection Method | Ref. |
|---|---|---|---|---|---|---|---|
| TRPV1 (VR1)(h) (antagonist effect) | human recombinant (CHO cells) | capsaicin (30 nM) | capsazepine | intracellular [Ca2+] | RT | Fluorimetry | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andres-Mach, M.; Zagaja, M.; Szala-Rycaj, J.; Szewczyk, A.; Abram, M.; Jakubiec, M.; Ciepiela, K.; Socała, K.; Wlaź, P.; Latacz, G.; et al. In Vivo and In Vitro Characterization of Close Analogs of Compound KA-11, a New Antiseizure Drug Candidate. Int. J. Mol. Sci. 2023, 24, 8302. https://doi.org/10.3390/ijms24098302
Andres-Mach M, Zagaja M, Szala-Rycaj J, Szewczyk A, Abram M, Jakubiec M, Ciepiela K, Socała K, Wlaź P, Latacz G, et al. In Vivo and In Vitro Characterization of Close Analogs of Compound KA-11, a New Antiseizure Drug Candidate. International Journal of Molecular Sciences. 2023; 24(9):8302. https://doi.org/10.3390/ijms24098302
Chicago/Turabian StyleAndres-Mach, Marta, Mirosław Zagaja, Joanna Szala-Rycaj, Aleksandra Szewczyk, Michał Abram, Marcin Jakubiec, Katarzyna Ciepiela, Katarzyna Socała, Piotr Wlaź, Gniewomir Latacz, and et al. 2023. "In Vivo and In Vitro Characterization of Close Analogs of Compound KA-11, a New Antiseizure Drug Candidate" International Journal of Molecular Sciences 24, no. 9: 8302. https://doi.org/10.3390/ijms24098302
APA StyleAndres-Mach, M., Zagaja, M., Szala-Rycaj, J., Szewczyk, A., Abram, M., Jakubiec, M., Ciepiela, K., Socała, K., Wlaź, P., Latacz, G., Khan, N., & Kaminski, K. (2023). In Vivo and In Vitro Characterization of Close Analogs of Compound KA-11, a New Antiseizure Drug Candidate. International Journal of Molecular Sciences, 24(9), 8302. https://doi.org/10.3390/ijms24098302