
From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID

 , , , , , , , , , , ,
, , , , , , , , , , ,  and
and
Abstract
1. Introduction
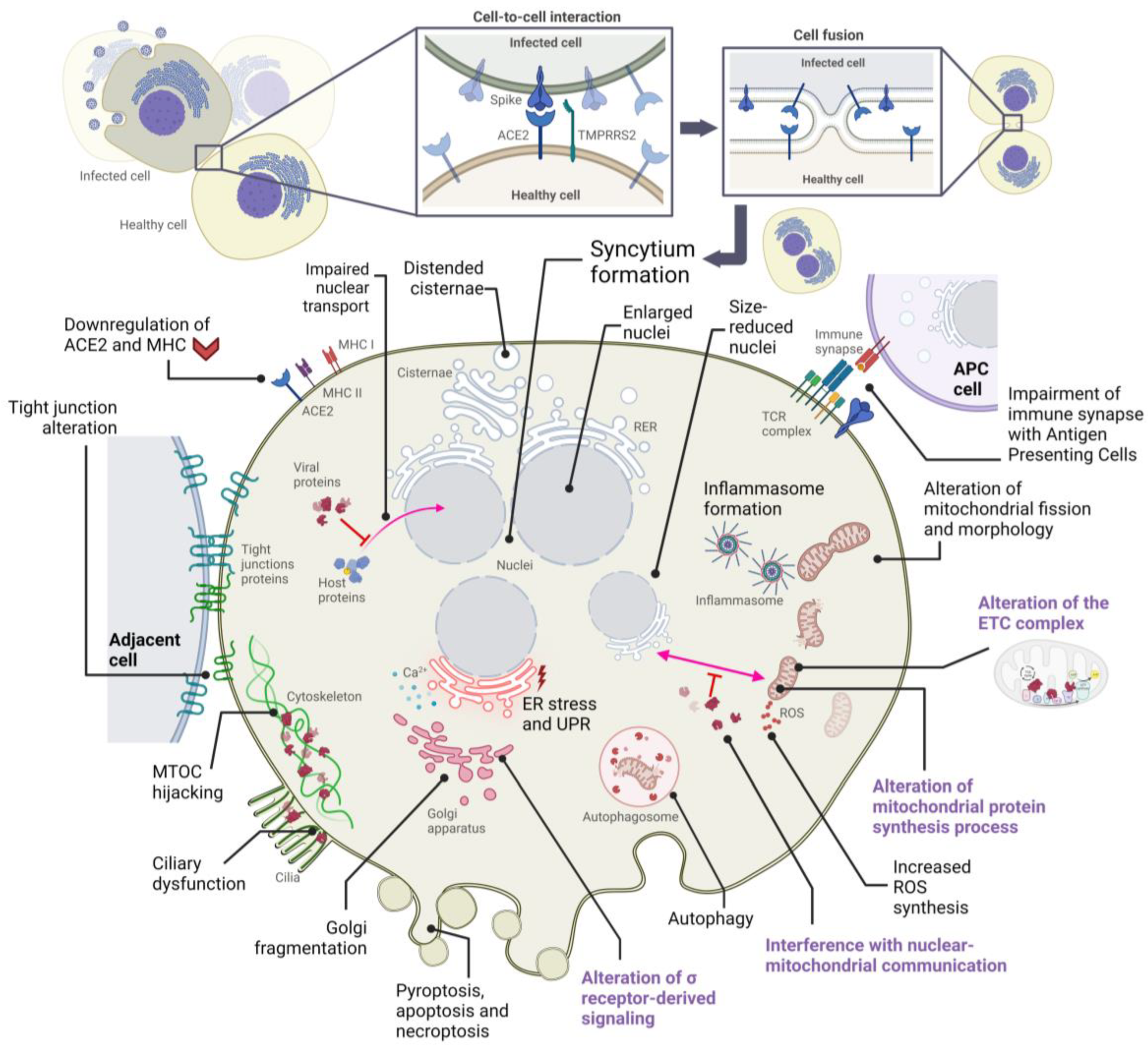
2. Alterations in Cellular Structures and Organelles Due to SARS-CoV-2 Infection
2.1. Cytopathic Effects on Mitochondria
2.2. Cytopathic Effects on the Endoplasmic Reticulum
2.3. Cytopathic Effects on the Golgi Apparatus
2.4. Cytopathic Effects on the Cytoskeleton and Plasma Membrane
2.5. Cytopathic Effects on the Nucleus
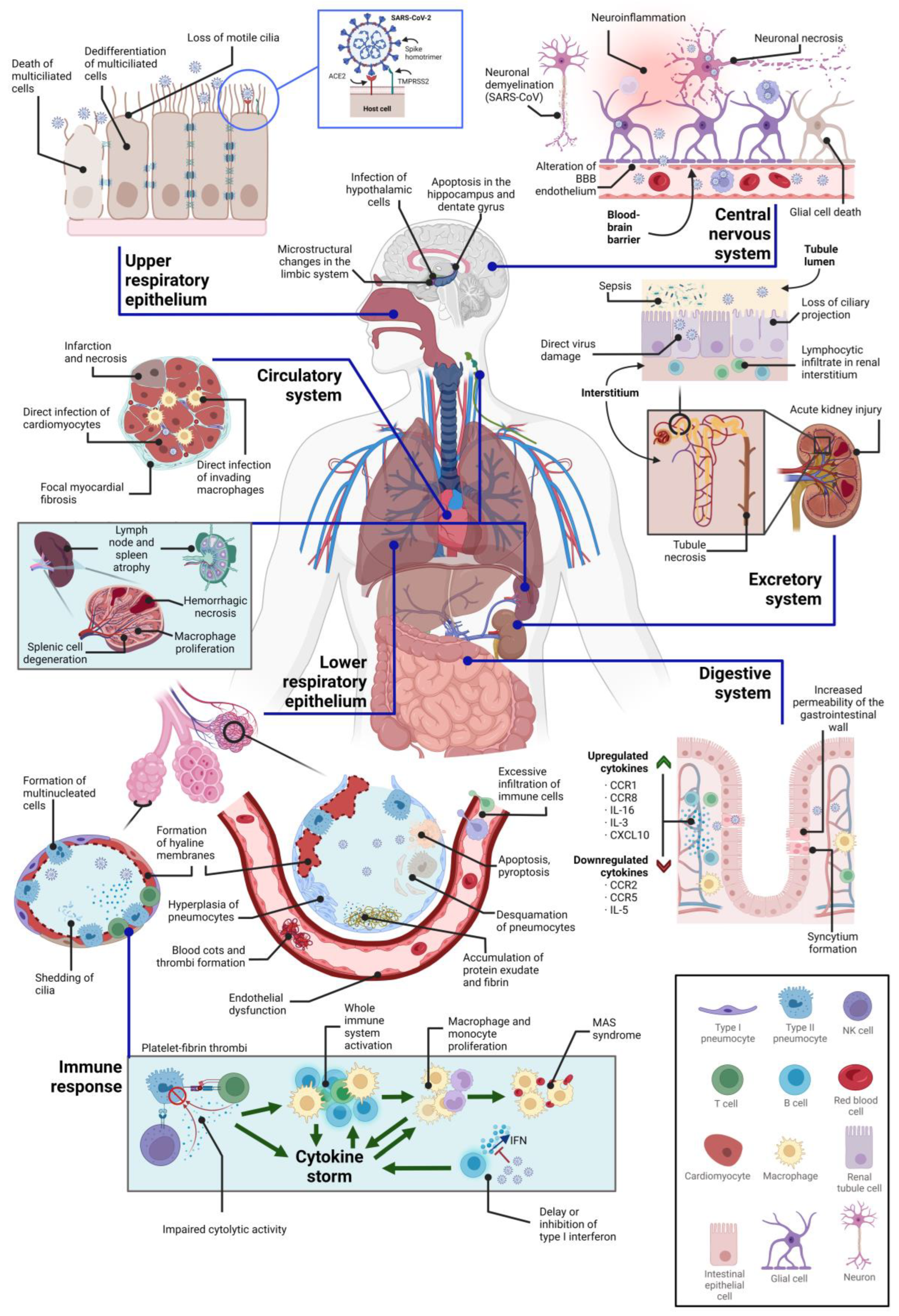
3. Direct Cytopathic Effects on Various Tissues
3.1. Cytopathic Effects on the Central Nervous System
3.2. Cytopathic Effects on the Respiratory System
3.3. Cytopathic Effects on the Circulatory System
3.4. Cytopathic Effects on the Immune System
3.5. Cytopathic Effects on the Kidney
3.6. Cytopathic Effects on the Digestive System
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-NCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The Molecular Virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Gramberg, T.; Simmons, G.; Möller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR Interact with the Glycoprotein of Marburg Virus and the S Protein of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Wong, A.H.M.; Zhou, D.; Rini, J.M. The X-ray Crystal Structure of Human Aminopeptidase N Reveals a Novel Dimer and the Basis for Peptide Processing. J. Biol. Chem. 2012, 287, 36804–36813. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, P.; Wang, P.; Li, Y.; Jiang, L.; Jia, W.; Wang, H.; Fan, A.; Wang, D.; Shi, X.; et al. Structural Definition of a Unique Neutralization Epitope on the Receptor-Binding Domain of MERS-CoV Spike Glycoprotein. Cell Rep. 2018, 24, 441–452. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Zelus, B.D.; Schickli, J.H.; Blau, D.M.; Weiss, S.R.; Holmes, K.V. Conformational Changes in the Spike Glycoprotein of Murine Coronavirus Are Induced at 37 °C Either by Soluble Murine CEACAM1 Receptors or by PH 8. J. Virol. 2003, 77, 830–840. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Banach, M.; Reiner, Ž.; Pirro, M.; Bianconi, V.; Al-Rasadi, K.; Sahebkar, A. Interaction Between Coronavirus S-Protein and Human ACE2: Hints for Exploring Efficient Therapeutic Targets to Treat COVID-19. Angiology 2021, 72, 122–130. [Google Scholar] [CrossRef]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of Ferrets, Cats, Dogs, and Other Domesticated Animals to SARS-Coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence That TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 Infects Cells after Viral Entry via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 Envelope and Membrane Proteins Modulate Maturation and Retention of the Spike Protein, Allowing Assembly of Virus-like Particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Khan, M.T.; Irfan, M.; Ahsan, H.; Ahmed, A.; Kaushik, A.C.; Khan, A.S.; Chinnasamy, S.; Ali, A.; Wei, D.-Q. Structures of SARS-CoV-2 RNA-Binding Proteins and Therapeutic Targets. Intervirology 2021, 64, 55–68. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Brian, D.A. Subgenomic Messenger RNA Amplification in Coronaviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12257–12262. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 Tissue Atlases Reveal SARS-CoV-2 Pathology and Cellular Targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-M.; Mannan, R.; Xiao, L.; Abdulfatah, E.; Qiao, Y.; Farver, C.; Myers, J.L.; Zelenka-Wang, S.; McMurry, L.; Su, F.; et al. Characterization of SARS-CoV-2 and Host Entry Factors Distribution in a COVID-19 Autopsy Series. Commun. Med. 2021, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major Findings, Mechanisms and Recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-Acute COVID-19 Syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Kozlov, A.V.; Camara, A.K.S. Mitochondria in Health and Diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef]
- Nunn, A.V.W.; Guy, G.W.; Brysch, W.; Bell, J.D. Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems. Biomedicines 2022, 10, 3113. [Google Scholar] [CrossRef]
- Morita, M.; Ler, L.W.; Fabian, M.R.; Siddiqui, N.; Mullin, M.; Henderson, V.C.; Alain, T.; Fonseca, B.D.; Karashchuk, G.; Bennett, C.F.; et al. A Novel 4EHP-GIGYF2 Translational Repressor Complex Is Essential for Mammalian Development. Mol. Cell. Biol. 2012, 32, 3585–3593. [Google Scholar] [CrossRef]
- Zhao, G.; Shi, S.-Q.; Yang, Y.; Peng, J.-P. M and N Proteins of SARS Coronavirus Induce Apoptosis in HPF Cells. Cell Biol. Toxicol. 2006, 22, 313–322. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, L. ACE2 Partially Dictates the Host Range and Tropism of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2020, 18, 4040–4047. [Google Scholar] [CrossRef]
- Archer, S.L.; Dasgupta, A.; Chen, K.-H.; Wu, D.; Baid, K.; Mamatis, J.E.; Gonzalez, V.; Read, A.; Bentley, R.E.; Martin, A.Y.; et al. SARS-CoV-2 Mitochondriopathy in COVID-19 Pneumonia Exacerbates Hypoxemia. Redox Biol. 2022, 58, 102508. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Walter, M.; Chen, I.P.; Vallejo-Gracia, A.; Kim, I.-J.; Bielska, O.; Lam, V.L.; Hayashi, J.M.; Cruz, A.; Shah, S.; Soveg, F.W.; et al. SIRT5 Is a Proviral Factor That Interacts with SARS-CoV-2 Nsp14 Protein. PLoS Pathog. 2022, 18, e1010811. [Google Scholar] [CrossRef]
- Batra, N.; De Souza, C.; Batra, J.; Raetz, A.G.; Yu, A.-M. The HMOX1 Pathway as a Promising Target for the Treatment and Prevention of SARS-CoV-2 of 2019 (COVID-19). Int. J. Mol. Sci. 2020, 21, 6412. [Google Scholar] [CrossRef]
- Wang, T.; Cao, Y.; Zhang, H.; Wang, Z.; Man, C.H.; Yang, Y.; Chen, L.; Xu, S.; Yan, X.; Zheng, Q.; et al. COVID-19 Metabolism: Mechanisms and Therapeutic Targets. MedComm 2022, 3, e157. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Neupane, N.; Rajendran, J.; Kvist, J.; Harjuhaahto, S.; Hu, B.; Kinnunen, V.; Yang, Y.; Nieminen, A.I.; Tyynismaa, H. Inter-Organellar and Systemic Responses to Impaired Mitochondrial Matrix Protein Import in Skeletal Muscle. Commun. Biol. 2022, 5, 1060. [Google Scholar] [CrossRef]
- Jiang, H.-W.; Zhang, H.-N.; Meng, Q.-F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.-X.; Wang, X.-N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b Suppresses Type I Interferon Responses by Targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, C.E.; Wooldredge, A.C.; Fong, B.; Kennedy, B.K.; Zhou, C. Tom70-Based Transcriptional Regulation of Mitochondrial Biogenesis and Aging. eLife 2022, 11, e75658. [Google Scholar] [CrossRef]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus Interactions with the Cellular Autophagy Machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef]
- Du, C.; Liu, W.-J.; Yang, J.; Zhao, S.-S.; Liu, H.-X. The Role of Branched-Chain Amino Acids and Branched-Chain α-Keto Acid Dehydrogenase Kinase in Metabolic Disorders. Front. Nutr. 2022, 9, 932670. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Wang, L.; Aliyari, S.; Cheng, G. SARS-CoV-2 Virus NSP14 Impairs NRF2/HMOX1 Activation by Targeting Sirtuin 1. Cell. Mol. Immunol. 2022, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Fernandes, L.; Lazari, L.C.; da Silva, J.M.; de Morais Gomes, V.; Machado, R.R.G.; dos Santos, A.F.; Araujo, D.B.; Coutinho, J.V.P.; Arini, G.S.; Angeli, C.B.; et al. SARS-CoV-2 Activates ER Stress and Unfolded Protein Response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Aoe, T. Pathological Aspects of COVID-19 as a Conformational Disease and the Use of Pharmacological Chaperones as a Potential Therapeutic Strategy. Front. Pharmacol. 2020, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic Reticulum as a Potential Therapeutic Target for COVID-19 Infection Management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Gupta, S. Endoplasmic Reticulum Secretory Pathway: Potential Target against SARS-CoV-2. Virus Res. 2022, 320, 198897. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 Membrane Protein Essential for Virus Assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef]
- Rashid, F.; Dzakah, E.E.; Wang, H.; Tang, S. The ORF8 Protein of SARS-CoV-2 Induced Endoplasmic Reticulum Stress and Mediated Immune Evasion by Antagonizing Production of Interferon Beta. Virus Res. 2021, 296, 198350. [Google Scholar] [CrossRef]
- Yao, L.; Xie, D.; Geng, L.; Shi, D.; Huang, J.; Wu, Y.; Lv, F.; Liang, D.; Li, L.; Liu, Y.; et al. REEP5 (Receptor Accessory Protein 5) Acts as a Sarcoplasmic Reticulum Membrane Sculptor to Modulate Cardiac Function. J. Am. Heart Assoc. 2018, 7, e007205. [Google Scholar] [CrossRef]
- Björk, S.; Hurt, C.M.; Ho, V.K.; Angelotti, T. REEPs Are Membrane Shaping Adapter Proteins That Modulate Specific G Protein-Coupled Receptor Trafficking by Affecting ER Cargo Capacity. PLoS ONE 2013, 8, e76366. [Google Scholar] [CrossRef]
- Son, Y.; Choi, C.; Saha, A.; Park, J.-H.; Im, H.; Cho, Y.K.; Seong, J.K.; Burl, R.B.; Rondini, E.A.; Granneman, J.G.; et al. REEP6 Knockout Leads to Defective β-Adrenergic Signaling in Adipocytes and Promotes Obesity-Related Metabolic Dysfunction. Metabolism 2022, 130, 155159. [Google Scholar] [CrossRef]
- Feng, L.; Yin, Y.-Y.; Liu, C.-H.; Xu, K.-R.; Li, Q.-R.; Wu, J.-R.; Zeng, R. Proteome-Wide Data Analysis Reveals Tissue-Specific Network Associated with SARS-CoV-2 Infection. J. Mol. Cell Biol. 2021, 12, 946–957. [Google Scholar] [CrossRef]
- Park, C.R.; You, D.-J.; Park, S.; Mander, S.; Jang, D.-E.; Yeom, S.-C.; Oh, S.-H.; Ahn, C.; Lee, S.H.; Seong, J.Y.; et al. The Accessory Proteins REEP5 and REEP6 Refine CXCR1-Mediated Cellular Responses and Lung Cancer Progression. Sci. Rep. 2016, 6, 39041. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R.A.J.O. Potential Applications for Sigma Receptor Ligands in Cancer Diagnosis and Therapy. Biochim. Biophys. Acta BBA-Biomembr. 2015, 1848, 2703–2714. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Lu, H.-L.; Zhang, L.-J.; Wu, Z. Sigma-2 Receptor Ligands and Their Perspectives in Cancer Diagnosis and Therapy: Sigma-2 Receptor Ligands. Med. Res. Rev. 2014, 34, 532–566. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernández-Castañeda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the Sigma-1 Receptor–IRE1 Pathway Is Beneficial in Preclinical Models of Inflammation and Sepsis. Sci. Transl. Med. 2019, 11, eaau5266. [Google Scholar] [CrossRef]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the Gene That Codes for the σ2 Receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef]
- Ahmed, I.S.A.; Chamberlain, C.; Craven, R.J. S2R Pgrmc1: The Cytochrome-Related Sigma-2 Receptor That Regulates Lipid and Drug Metabolism and Hormone Signaling. Expert Opin. Drug Metab. Toxicol. 2012, 8, 361–370. [Google Scholar] [CrossRef]
- Skuza, G. Potential Antidepressant Activity of Sigma Ligands. Pol. J. Pharmacol. 2003, 55, 923–934. [Google Scholar]
- Tang, S.W.; Leonard, B.E.; Helmeste, D.M. Long COVID, Neuropsychiatric Disorders, Psychotropics, Present and Future. Acta Neuropsychiatr. 2022, 34, 109–126. [Google Scholar] [CrossRef]
- Hashimoto, K. Repurposing of CNS Drugs to Treat COVID-19 Infection: Targeting the Sigma-1 Receptor. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Sun, Y.; Diaz-Ruiz, A.; Ali, A.; Gutierrez, V.; Palacios, H.H.; Curtis, J.; Siendones, E.; Ariza, J.; Abulwerdi, G.A.; et al. Cytochrome B5 Reductase and the Control of Lipid Metabolism and Healthspan. NPJ Aging Mech. Dis. 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, M.; Kanzaki, M.; Iino, Y.; Morishita, Y.; Kojima, I. Identification of a Novel Chloride Channel Expressed in the Endoplasmic Reticulum, Golgi Apparatus, and Nucleus. J. Biol. Chem. 2001, 276, 20413–20418. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yoo, Y.; Fan, H.; Kim, E.; Guan, K.-L.; Guan, J.-L. Regulation of Integrin β 1 Recycling to Lipid Rafts by Rab1a to Promote Cell Migration. J. Biol. Chem. 2010, 285, 29398–29405. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.M.; Molinari, M. Coronaviruses Hijack the LC3-I-Positive EDEMosomes, ER-Derived Vesicles Exporting Short-Lived ERAD Regulators, for Replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef]
- Sicari, D.; Chatziioannou, A.; Koutsandreas, T.; Sitia, R.; Chevet, E. Role of the Early Secretory Pathway in SARS-CoV-2 Infection. J. Cell Biol. 2020, 219, e202006005. [Google Scholar] [CrossRef]
- Yiang, G.-T.; Wu, C.-C.; Lu, C.-L.; Hu, W.-C.; Tsai, Y.-J.; Huang, Y.-M.; Su, W.-L.; Lu, K.-C. Endoplasmic Reticulum Stress in Elderly Patients with COVID-19: Potential of Melatonin Treatment. Viruses 2023, 15, 156. [Google Scholar] [CrossRef]
- Cortese, M.; Lee, J.-Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e5. [Google Scholar] [CrossRef]
- Zhang, J.; Kennedy, A.; Xing, L.; Bui, S.; Reid, W.; Joppich, J.; Ahat, E.; Rose, M.; Tang, Q.; Tai, A.W.; et al. SARS-CoV-2 Triggers Golgi Fragmentation via down-Regulation of GRASP55 to Facilitate Viral Trafficking. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Li, T.; Jiang, Z.; Zeng, L.; Hu, Z. The Role of the Golgi Apparatus in Disease (Review). Int. J. Mol. Med. 2021, 47, 38. [Google Scholar] [CrossRef]
- Wang, Y.; Gandy, S. The Golgi Apparatus: Site for Convergence of COVID-19 Brain Fog and Alzheimer’s Disease? Mol. Neurodegener. 2022, 17, 67. [Google Scholar] [CrossRef]
- Devergnas, S.; Chimienti, F.; Naud, N.; Pennequin, A.; Coquerel, Y.; Chantegrel, J.; Favier, A.; Seve, M. Differential Regulation of Zinc Efflux Transporters ZnT-1, ZnT-5 and ZnT-7 Gene Expression by Zinc Levels: A Real-Time RT-PCR Study. Biochem. Pharmacol. 2004, 68, 699–709. [Google Scholar] [CrossRef]
- Kirschke, C.P.; Huang, L. ZnT7, a Novel Mammalian Zinc Transporter, Accumulates Zinc in the Golgi Apparatus. J. Biol. Chem. 2003, 278, 4096–4102. [Google Scholar] [CrossRef]
- Matern, H.; Yang, X.; Andrulis, E.; Sternglanz, R.; Trepte, H.H.; Gallwitz, D. A Novel Golgi Membrane Protein Is Part of a GTPase-Binding Protein Complex Involved in Vesicle Targeting. EMBO J. 2000, 19, 4485–4492. [Google Scholar] [CrossRef]
- Schulz, J.; Avci, D.; Queisser, M.A.; Gutschmidt, A.; Dreher, L.-S.; Fenech, E.J.; Volkmar, N.; Hayashi, Y.; Hoppe, T.; Christianson, J.C. Conserved Cytoplasmic Domains Promote Hrd1 Ubiquitin Ligase Complex Formation for ER-Associated Degradation (ERAD). J. Cell Sci. 2017, 130, 3322–3335. [Google Scholar] [CrossRef]
- van de Weijer, M.L.; Krshnan, L.; Liberatori, S.; Guerrero, E.N.; Robson-Tull, J.; Hahn, L.; Lebbink, R.J.; Wiertz, E.J.H.J.; Fischer, R.; Ebner, D.; et al. Quality Control of ER Membrane Proteins by the RNF185/Membralin Ubiquitin Ligase Complex. Mol. Cell 2020, 79, 768–781.e7. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, Y.; Zhu, H.; Ahmed, K.; Fu, C.; Yao, X. Human Yip1A Specifies the Localization of Yif1 to the Golgi Apparatus. Biochem. Biophys. Res. Commun. 2005, 334, 16–22. [Google Scholar] [CrossRef]
- Adelino, J.E.; Addobbati, C.; Pontillo, A.; Fragoso, T.S.; Duarte, Â.; Crovella, S.; De Azevedo Silva, J.; Sandrin-Garcia, P. A Genetic Variant within SLC30A6 Has a Protective Role in the Severity of Rheumatoid Arthritis. Scand. J. Rheumatol. 2017, 46, 326–327. [Google Scholar] [CrossRef]
- Fukunaka, A.; Suzuki, T.; Kurokawa, Y.; Yamazaki, T.; Fujiwara, N.; Ishihara, K.; Migaki, H.; Okumura, K.; Masuda, S.; Yamaguchi-Iwai, Y.; et al. Demonstration and Characterization of the Heterodimerization of ZnT5 and ZnT6 in the Early Secretory Pathway. J. Biol. Chem. 2009, 284, 30798–30806. [Google Scholar] [CrossRef]
- Wessels, I.; Rolles, B.; Rink, L. The Potential Impact of Zinc Supplementation on COVID-19 Pathogenesis. Front. Immunol. 2020, 11, 1712. [Google Scholar] [CrossRef]
- Mahmoud, M.M.; Abuohashish, H.M.; Khairy, D.A.; Bugshan, A.S.; Khan, A.M.; Moothedath, M.M. Pathogenesis of Dysgeusia in COVID-19 Patients: A Scoping Review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1114–1134. [Google Scholar] [CrossRef] [PubMed]
- Larocca, M.C.; Shanks, R.A.; Tian, L.; Nelson, D.L.; Stewart, D.M.; Goldenring, J.R. AKAP350 Interaction with Cdc42 Interacting Protein 4 at the Golgi Apparatus. Mol. Biol. Cell 2004, 15, 2771–2781. [Google Scholar] [CrossRef] [PubMed]
- Puthenveedu, M.A.; Bachert, C.; Puri, S.; Lanni, F.; Linstedt, A.D. GM130 and GRASP65-Dependent Lateral Cisternal Fusion Allows Uniform Golgi-Enzyme Distribution. Nat. Cell Biol. 2006, 8, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Witczak, O.; Skålhegg, B.S.; Keryer, G.; Bornens, M.; Taskén, K.; Jahnsen, T.; Orstavik, S. Cloning and Characterization of a CDNA Encoding an A-Kinase Anchoring Protein Located in the Centrosome, AKAP450. EMBO J. 1999, 18, 1858–1868. [Google Scholar] [CrossRef]
- Wu, J.; de Heus, C.; Liu, Q.; Bouchet, B.P.; Noordstra, I.; Jiang, K.; Hua, S.; Martin, M.; Yang, C.; Grigoriev, I.; et al. Molecular Pathway of Microtubule Organization at the Golgi Apparatus. Dev. Cell 2016, 39, 44–60. [Google Scholar] [CrossRef]
- Munro, S. The Golgin Coiled-Coil Proteins of the Golgi Apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a005256. [Google Scholar] [CrossRef]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Weiss, R.J.; Spahn, P.N.; Toledo, A.G.; Chiang, A.W.T.; Kellman, B.P.; Li, J.; Benner, C.; Glass, C.K.; Gordts, P.L.S.M.; Lewis, N.E.; et al. ZNF263 Is a Transcriptional Regulator of Heparin and Heparan Sulfate Biosynthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 9311–9317. [Google Scholar] [CrossRef]
- Kloc, M.; Uosef, A.; Wosik, J.; Kubiak, J.Z.; Ghobrial, R.M. Virus Interactions with the Actin Cytoskeleton—What We Know and Do Not Know about SARS-CoV-2. Arch. Virol. 2022, 167, 737–749. [Google Scholar] [CrossRef]
- Aminpour, M.; Hameroff, S.; Tuszynski, J.A. How COVID-19 Hijacks the Cytoskeleton: Therapeutic Implications. Life 2022, 12, 814. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Oldridge, D.A.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep Immune Profiling of COVID-19 Patients Reveals Patient Heterogeneity and Distinct Immunotypes with Implications for Therapeutic Interventions. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Michie, K.A.; Bermeister, A.; Robertson, N.O.; Goodchild, S.C.; Curmi, P.M.G. Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int. J. Mol. Sci. 2019, 20, 1996. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Heissler, S.M.; Eto, M.; Nishimura, Y.; Fischer, R.S.; Thiam, H.R.; Waterman, C.M. MARK2 Regulates Directed Cell Migration through Modulation of Myosin II Contractility and Focal Adhesion Organization. Curr. Biol. 2022, 32, 2704–2718.e6. [Google Scholar] [CrossRef]
- Thies, E.; Mandelkow, E.-M. Missorting of Tau in Neurons Causes Degeneration of Synapses That Can Be Rescued by the Kinase MARK2/Par-1. J. Neurosci. 2007, 27, 2896–2907. [Google Scholar] [CrossRef]
- Matenia, D.; Hempp, C.; Timm, T.; Eikhof, A.; Mandelkow, E.-M. Microtubule Affinity-Regulating Kinase 2 (MARK2) Turns on Phosphatase and Tensin Homolog (PTEN)-Induced Kinase 1 (PINK1) at Thr-313, a Mutation Site in Parkinson Disease. J. Biol. Chem. 2012, 287, 8174–8186. [Google Scholar] [CrossRef]
- Pera, T.; Tompkins, E.; Katz, M.; Wang, B.; Deshpande, D.A.; Weinman, E.J.; Penn, R.B. Specificity of NHERF1 Regulation of GPCR Signaling and Function in Human Airway Smooth Muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9008–9016. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of MRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Szymanski, D. Tubulin Folding Cofactors: Half a Dozen for a Dimer. Curr. Biol. CB 2002, 12, R767–R769. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dona, M.; Hetterschijt, L.; Tonnaer, E.; Peters, T.; de Vrieze, E.; Mans, D.A.; van Beersum, S.E.C.; Phelps, I.G.; Arts, H.H.; et al. The Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking. PLoS Genet. 2015, 11, e1005575. [Google Scholar] [CrossRef]
- Dona, M.; Bachmann-Gagescu, R.; Texier, Y.; Toedt, G.; Hetterschijt, L.; Tonnaer, E.L.; Peters, T.A.; van Beersum, S.E.C.; Bergboer, J.G.M.; Horn, N.; et al. NINL and DZANK1 Co-Function in Vesicle Transport and Are Essential for Photoreceptor Development in Zebrafish. PLoS Genet. 2015, 11, e1005574. [Google Scholar] [CrossRef]
- van Wijk, E.; Kersten, F.F.J.; Kartono, A.; Mans, D.A.; Brandwijk, K.; Letteboer, S.J.F.; Peters, T.A.; Märker, T.; Yan, X.; Cremers, C.W.R.J.; et al. Usher Syndrome and Leber Congenital Amaurosis Are Molecularly Linked via a Novel Isoform of the Centrosomal Ninein-like Protein. Hum. Mol. Genet. 2009, 18, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, C.; Yang, B.; Zhang, H.; Jiao, J.; Zhang, R.; Liu, S.; Xiao, S.; Chen, Y.; Liu, B.; et al. SARS-CoV-2 ORF10 Impairs Cilia by Enhancing CUL2ZYG11B Activity. J. Cell Biol. 2022, 221, e202108015. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Asfahani, R.; Carroll, P.; Bicknell, L.; Lescai, F.; Bright, A.; Chanudet, E.; Brooks, A.; Christou-Savina, S.; Osman, G.; et al. The Kinetochore Protein, CENPF, Is Mutated in Human Ciliopathy and Microcephaly Phenotypes. J. Med. Genet. 2015, 52, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A. Airway Epithelial Differentiation and Mucociliary Clearance. Ann. Am. Thorac. Soc. 2018, 15, S143–S148. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.A.; Mitsopoulos, P.; Blagih, J.; Dunn, S.D.; St-Pierre, J.; Jones, R.G.; Hatch, G.M.; Madrenas, J. Stomatin-like Protein 2 Deficiency in T Cells Is Associated with Altered Mitochondrial Respiration and Defective CD4+ T Cell Responses. J. Immunol. 2012, 189, 4349–4360. [Google Scholar] [CrossRef]
- Onnis, A.; Andreano, E.; Cassioli, C.; Finetti, F.; Della Bella, C.; Staufer, O.; Pantano, E.; Abbiento, V.; Marotta, G.; D’Elios, M.M.; et al. SARS-CoV-2 Spike Protein Suppresses CTL-Mediated Killing by Inhibiting Immune Synapse Assembly. J. Exp. Med. 2023, 220, e20220906. [Google Scholar] [CrossRef]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the Immunological Synapse: A Key to HIV-1 Pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef]
- Abdel Hameid, R.; Cormet-Boyaka, E.; Kuebler, W.M.; Uddin, M.; Berdiev, B.K. SARS-CoV-2 May Hijack GPCR Signaling Pathways to Dysregulate Lung Ion and Fluid Transport. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 320, L430–L435. [Google Scholar] [CrossRef]
- Motley, A.; Bright, N.A.; Seaman, M.N.J.; Robinson, M.S. Clathrin-Mediated Endocytosis in AP-2-Depleted Cells. J. Cell Biol. 2003, 162, 909–918. [Google Scholar] [CrossRef]
- Liu, Q.; Bautista-Gomez, J.; Higgins, D.A.; Yu, J.; Xiong, Y. Dysregulation of the AP2M1 Phosphorylation Cycle by LRRK2 Impairs Endocytosis and Leads to Dopaminergic Neurodegeneration. Sci. Signal. 2021, 14, eabg3555. [Google Scholar] [CrossRef]
- Karim, M.; Saul, S.; Ghita, L.; Sahoo, M.K.; Ye, C.; Bhalla, N.; Lo, C.-W.; Jin, J.; Park, J.-G.; Martinez-Gualda, B.; et al. Numb-Associated Kinases Are Required for SARS-CoV-2 Infection and Are Cellular Targets for Antiviral Strategies. Antivir. Res. 2022, 204, 105367. [Google Scholar] [CrossRef]
- Puray-Chavez, M.; LaPak, K.M.; Schrank, T.P.; Elliott, J.L.; Bhatt, D.P.; Agajanian, M.J.; Jasuja, R.; Lawson, D.Q.; Davis, K.; Rothlauf, P.W.; et al. Systematic Analysis of SARS-CoV-2 Infection of an ACE2-Negative Human Airway Cell. Cell Rep. 2021, 36, 109364. [Google Scholar] [CrossRef]
- Schreiner, T.; Allnoch, L.; Beythien, G.; Marek, K.; Becker, K.; Schaudien, D.; Stanelle-Bertram, S.; Schaumburg, B.; Mounogou Kouassi, N.; Beck, S.; et al. SARS-CoV-2 Infection Dysregulates Cilia and Basal Cell Homeostasis in the Respiratory Epithelium of Hamsters. Int. J. Mol. Sci. 2022, 23, 5124. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 Nsp12 Attenuates Type I Interferon Production by Inhibiting IRF3 Nuclear Translocation. Cell. Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Kato, K.; Ikliptikawati, D.K.; Kobayashi, A.; Kondo, H.; Lim, K.; Hazawa, M.; Wong, R.W. Overexpression of SARS-CoV-2 Protein ORF6 Dislocates RAE1 and NUP98 from the Nuclear Pore Complex. Biochem. Biophys. Res. Commun. 2021, 536, 59–66. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 Hijacks Nup98 to Block STAT Nuclear Import and Antagonize Interferon Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N Protein Antagonizes Type I Interferon Signaling by Suppressing Phosphorylation and Nuclear Translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 65. [Google Scholar] [CrossRef]
- Collins, S.E.; Noyce, R.S.; Mossman, K.L. Innate Cellular Response to Virus Particle Entry Requires IRF3 but Not Virus Replication. J. Virol. 2004, 78, 1706–1717. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 Protein of SARS-CoV-2 Disrupts the MRNA Export Machinery to Inhibit Host Gene Expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Matuck, B.F.; Dolhnikoff, M.; Duarte-Neto, A.N.; Maia, G.; Gomes, S.C.; Sendyk, D.I.; Zarpellon, A.; de Andrade, N.P.; Monteiro, R.A.; Pinho, J.R.R.; et al. Salivary Glands Are a Target for SARS-CoV-2: A Source for Saliva Contamination. J. Pathol. 2021, 254, 239–243. [Google Scholar] [CrossRef]
- Nardacci, R.; Colavita, F.; Castilletti, C.; Lapa, D.; Matusali, G.; Meschi, S.; Del Nonno, F.; Colombo, D.; Capobianchi, M.R.; Zumla, A.; et al. Evidences for Lipid Involvement in SARS-CoV-2 Cytopathogenesis. Cell Death Dis. 2021, 12, 263. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia Formation by SARS-CoV-2-Infected Cells. EMBO J. 2021, 40, e107405. [Google Scholar] [CrossRef] [PubMed]
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of Viral RNA, Pneumocyte Syncytia and Thrombosis Are Hallmarks of Advanced COVID-19 Pathology. EBioMedicine 2020, 61, 103104. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Med. Kaunas Lith. 2021, 58, 16. [Google Scholar] [CrossRef] [PubMed]
- LeGros, H.L.; Halim, A.B.; Geller, A.M.; Kotb, M. Cloning, Expression, and Functional Characterization of the Beta Regulatory Subunit of Human Methionine Adenosyltransferase (MAT II). J. Biol. Chem. 2000, 275, 2359–2366. [Google Scholar] [CrossRef]
- Baig, A.M. Deleterious Outcomes in Long-Hauler COVID-19: The Effects of SARS-CoV-2 on the CNS in Chronic COVID Syndrome. ACS Chem. Neurosci. 2020, 11, 4017–4020. [Google Scholar] [CrossRef]
- Villadiego, J.; García-Arriaza, J.; Ramírez-Lorca, R.; García-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Álvarez-Vergara, M.I.; Cala-Fernández, F.; García-Roldán, E.; López-Ogáyar, J.L.; et al. Full Protection from SARS-CoV-2 Brain Infection and Damage in Susceptible Transgenic Mice Conferred by MVA-CoV2-S Vaccine Candidate. Nat. Neurosci. 2023, 26, 226–238. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Akerstrom, V. HIV-1 Protein Gp120 Crosses the Blood-Brain Barrier: Role of Adsorptive Endocytosis. Life Sci. 1997, 61, PL119–PL125. [Google Scholar] [CrossRef]
- Achar, A.; Ghosh, C. COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Relevance. Cells 2020, 9, 2360. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Benameur, K.; Agarwal, A.; Auld, S.C.; Butters, M.P.; Webster, A.S.; Ozturk, T.; Howell, J.C.; Bassit, L.C.; Velasquez, A.; Schinazi, R.F.; et al. Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020. Emerg. Infect. Dis. 2020, 26, 2016–2021. [Google Scholar] [CrossRef]
- Xia, H.; Lazartigues, E. Angiotensin-Converting Enzyme 2 in the Brain: Properties and Future Directions. J. Neurochem. 2008, 107, 1482–1494. [Google Scholar] [CrossRef]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a New Potential SARS-CoV-2 Infection Mediator Implicated in the Neurologic Features and Central Nervous System Involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef]
- Solomon, T. Neurological Infection with SARS-CoV-2—The Story so Far. Nat. Rev. Neurol. 2021, 17, 65–66. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; Troakes, C.; Hanley, B.; Osborn, M.; Richardson, M.P.; Hotopf, M.; Bullmore, E.; Everall, I.P. Invited Review: The Spectrum of Neuropathology in COVID-19. Neuropathol. Appl. Neurobiol. 2021, 47, 3–16. [Google Scholar] [CrossRef]
- Desai, A.D.; Lavelle, M.; Boursiquot, B.C.; Wan, E.Y. Long-Term Complications of COVID-19. Am. J. Physiol. Cell Physiol. 2022, 322, C1–C11. [Google Scholar] [CrossRef]
- Visco, V.; Vitale, C.; Rispoli, A.; Izzo, C.; Virtuoso, N.; Ferruzzi, G.J.; Santopietro, M.; Melfi, A.; Rusciano, M.R.; Maglio, A.; et al. Post-COVID-19 Syndrome: Involvement and Interactions between Respiratory, Cardiovascular and Nervous Systems. J. Clin. Med. 2022, 11, 524. [Google Scholar] [CrossRef]
- Hugon, J.; Msika, E.-F.; Queneau, M.; Farid, K.; Paquet, C. Long COVID: Cognitive Complaints (Brain Fog) and Dysfunction of the Cingulate Cortex. J. Neurol. 2022, 269, 44–46. [Google Scholar] [CrossRef]
- Backman, L.; Möller, M.C.; Thelin, E.P.; Dahlgren, D.; Deboussard, C.; Östlund, G.; Lindau, M. Monthlong Intubated Patient with Life-Threatening COVID-19 and Cerebral Microbleeds Suffers Only Mild Cognitive Sequelae at 8-Month Follow-up: A Case Report. Arch. Clin. Neuropsychol. Off. J. Natl. Acad. Neuropsychol. 2022, 37, 531–543. [Google Scholar] [CrossRef]
- Nau, R.; Soto, A.; Bruck, W. Apoptosis of Neurons in the Dentate Gyrus in Humans Suffering from Bacterial Meningitis. J. Neuropathol. Exp. Neurol. 1999, 58, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 Main Protease Mpro Causes Microvascular Brain Pathology by Cleaving NEMO in Brain Endothelial Cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Salzano, C.; Saracino, G.; Cardillo, G. Possible Adrenal Involvement in Long COVID Syndrome. Medicina 2021, 57, 1087. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Gubbi, S.; Koch, C.A. COVID-19 and Chronic Fatigue Syndrome: An Endocrine Perspective. J. Clin. Transl. Endocrinol. 2022, 27, 100284. [Google Scholar] [CrossRef]
- Qin, Y.; Wu, J.; Chen, T.; Li, J.; Zhang, G.; Wu, D.; Zhou, Y.; Zheng, N.; Cai, A.; Ning, Q.; et al. Long-Term Microstructure and Cerebral Blood Flow Changes in Patients Recovered from COVID-19 without Neurological Manifestations. J. Clin. Investig. 2021, 131, e147329. [Google Scholar] [CrossRef]
- Disser, N.P.; De Micheli, A.J.; Schonk, M.M.; Konnaris, M.A.; Piacentini, A.N.; Edon, D.L.; Toresdahl, B.G.; Rodeo, S.A.; Casey, E.K.; Mendias, C.L. Musculoskeletal Consequences of COVID-19. J. Bone Jt. Surg. 2020, 102, 1197–1204. [Google Scholar] [CrossRef]
- Wang, F.; Kream, R.M.; Stefano, G.B. Long-Term Respiratory and Neurological Sequelae of COVID-19. Med. Sci. Monit. 2020, 26, e928996-1. [Google Scholar] [CrossRef]
- Gallo, O.; Locatello, L.G.; Mazzoni, A.; Novelli, L.; Annunziato, F. The Central Role of the Nasal Microenvironment in the Transmission, Modulation, and Clinical Progression of SARS-CoV-2 Infection. Mucosal Immunol. 2021, 14, 305–316. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and Cytopathic Effect of SARS-CoV-2 Infection in Human Airway Epithelial Cells. Nat. Commun. 2020, 11, 3910. [Google Scholar] [CrossRef]
- Bridges, J.P.; Vladar, E.K.; Huang, H.; Mason, R.J. Respiratory Epithelial Cell Responses to SARS-CoV-2 in COVID-19. Thorax 2022, 77, 203–209. [Google Scholar] [CrossRef]
- Morrison, C.B.; Edwards, C.E.; Shaffer, K.M.; Araba, K.C.; Wykoff, J.A.; Williams, D.R.; Asakura, T.; Dang, H.; Morton, L.C.; Gilmore, R.C.; et al. SARS-CoV-2 Infection of Airway Cells Causes Intense Viral and Cell Shedding, Two Spreading Mechanisms Affected by IL-13. Proc. Natl. Acad. Sci. USA 2022, 119, e2119680119. [Google Scholar] [CrossRef]
- Takeda, K.; Sakakibara, S.; Yamashita, K.; Motooka, D.; Nakamura, S.; El Hussien, M.A.; Katayama, J.; Maeda, Y.; Nakata, M.; Hamada, S.; et al. Allergic Conversion of Protective Mucosal Immunity against Nasal Bacteria in Patients with Chronic Rhinosinusitis with Nasal Polyposis. J. Allergy Clin. Immunol. 2019, 143, 1163–1175.e15. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, J.; Hong, S.P.; Choi, S.Y.; Yang, M.J.; Ju, Y.S.; Kim, Y.T.; Kim, H.M.; Rahman, M.D.T.; Chung, M.K.; et al. Nasal Ciliated Cells Are Primary Targets for SARS-CoV-2 Replication in the Early Stage of COVID-19. J. Clin. Investig. 2021, 131, e148517. [Google Scholar] [CrossRef]
- Robinot, R.; Hubert, M.; de Melo, G.D.; Lazarini, F.; Bruel, T.; Smith, N.; Levallois, S.; Larrous, F.; Fernandes, J.; Gellenoncourt, S.; et al. SARS-CoV-2 Infection Induces the Dedifferentiation of Multiciliated Cells and Impairs Mucociliary Clearance. Nat. Commun. 2021, 12, 4354. [Google Scholar] [CrossRef]
- Wahl, A.; Gralinski, L.; Johnson, C.; Yao, W.; Kovarova, M.; Dinnon, K.; Liu, H.; Madden, V.; Krzystek, H.; De, C.; et al. Acute SARS-CoV-2 Infection Is Highly Cytopathic, Elicits a Robust Innate Immune Response and Is Efficiently Prevented by EIDD-2801. Res. Sq. 2020, rs.3.rs-80404. [Google Scholar] [CrossRef]
- Huang, B. Mucins Produced by Type II Pneumocyte: Culprits in SARS-CoV-2 Pathogenesis. Cell. Mol. Immunol. 2021, 18, 1823–1825. [Google Scholar] [CrossRef]
- Hu, G.; Christman, J.W. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front. Immunol. 2019, 10, 2275. [Google Scholar] [CrossRef]
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid Receptor Blocker Increases Angiotensin-Converting Enzyme 2 Activity in Congestive Heart Failure Patients. Circ. Res. 2005, 97, 946–953. [Google Scholar] [CrossRef]
- Gagnon, H.; Refaie, S.; Gagnon, S.; Desjardins, R.; Salzet, M.; Day, R. Proprotein Convertase 1/3 (PC1/3) in the Rat Alveolar Macrophage Cell Line NR8383: Localization, Trafficking and Effects on Cytokine Secretion. PLoS ONE 2013, 8, e61557. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, Y.; Li, K.; Meyerholz, D.K.; Allamargot, C.; Perlman, S. Severe Acute Respiratory Syndrome Coronavirus 2-Induced Immune Activation and Death of Monocyte-Derived Human Macrophages and Dendritic Cells. J. Infect. Dis. 2021, 223, 785–795. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Niles, M.A.; Gogesch, P.; Kronhart, S.; Ortega Iannazzo, S.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and Dendritic Cells Are Not the Major Source of Pro-Inflammatory Cytokines Upon SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 647824. [Google Scholar] [CrossRef] [PubMed]
- Carfì, A.; Bernabei, R.; Landi, F. Gemelli Against COVID-19 Post-Acute Care Study Group Persistent Symptoms in Patients after Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Castanares-Zapatero, D.; Chalon, P.; Kohn, L.; Dauvrin, M.; Detollenaere, J.; Maertens de Noordhout, C.; Primus-de Jong, C.; Cleemput, I.; Van den Heede, K. Pathophysiology and Mechanism of Long COVID: A Comprehensive Review. Ann. Med. 2022, 54, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Bernard, I.; Limonta, D.; Mahal, L.; Hobman, T. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Robson, B. Bioinformatics Studies on a Function of the SARS-CoV-2 Spike Glycoprotein as the Binding of Host Sialic Acid Glycans. Comput. Biol. Med. 2020, 122, 103849. [Google Scholar] [CrossRef]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef]
- Nader, D.; Fletcher, N.; Curley, G.F.; Kerrigan, S.W. SARS-CoV-2 Uses Major Endothelial Integrin Avβ3 to Cause Vascular Dysregulation in-Vitro during COVID-19. PLoS ONE 2021, 16, e0253347. [Google Scholar] [CrossRef]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and Derangement of Renin-Angiotensin-Aldosterone System in COVID-19: A Novel Hypothesis for Clinically Suspected Hypercoagulopathy and Microvascular Immunothrombosis. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 507, 167–173. [Google Scholar] [CrossRef]
- Costa, T.J.; Potje, S.R.; Fraga-Silva, T.F.C.; da Silva-Neto, J.A.; Barros, P.R.; Rodrigues, D.; Machado, M.R.; Martins, R.B.; Santos-Eichler, R.A.; Benatti, M.N.; et al. Mitochondrial DNA and TLR9 Activation Contribute to SARS-CoV-2-Induced Endothelial Cell Damage. Vascul. Pharmacol. 2022, 142, 106946. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE2. BioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Italia, L.; Tomasoni, D.; Bisegna, S.; Pancaldi, E.; Stretti, L.; Adamo, M.; Metra, M. COVID-19 and Heart Failure: From Epidemiology During the Pandemic to Myocardial Injury, Myocarditis, and Heart Failure Sequelae. Front. Cardiovasc. Med. 2021, 8, 713560. [Google Scholar] [CrossRef]
- Tudoran, C.; Tudoran, M.; Elena Lazureanu, V.; Raluca Marinescu, A.; Novacescu, D.; Georgiana Cut, T. Impairment of the Cardiovascular System during SARS-CoV-2 Infection. In RNA Viruses Infection; Shah, Y., Ed.; IntechOpen: London, UK, 2022; ISBN 978-1-80355-666-6. [Google Scholar]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Dixit, N.M.; Churchill, A.; Nsair, A.; Hsu, J.J. Post-Acute COVID-19 Syndrome and the Cardiovascular System: What Is Known? Am. Heart J. Plus Cardiol. Res. Pract. 2021, 5, 100025. [Google Scholar] [CrossRef]
- DePace, N.L.; Colombo, J. Long-COVID Syndrome and the Cardiovascular System: A Review of Neurocardiologic Effects on Multiple Systems. Curr. Cardiol. Rep. 2022, 24, 1711–1726. [Google Scholar] [CrossRef]
- Farshidfar, F.; Koleini, N.; Ardehali, H. Cardiovascular Complications of COVID-19. JCI Insight 2021, 6, e148980. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and Cardiovascular Disease: From Basic Mechanisms to Clinical Perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the Mystery Surrounding Post-Acute Sequelae of COVID-19. Front. Immunol. 2021, 12, 686029. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine Storm in COVID-19: Pathogenesis and Overview of Anti-Inflammatory Agents Used in Treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.; Gong, C.; Yan, X.; Si, G.; Fang, C.; Wang, L.; Zhu, X.; Xu, Z.; Yao, C.; Zhu, S. Targeting CD155 by Rediocide-A Overcomes Tumour Immuno-Resistance to Natural Killer Cells. Pharm. Biol. 2021, 59, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Paces, J.; Strizova, Z.; Smrz, D.; Cerny, J. COVID-19 and the Immune System. Physiol. Res. 2020, 69, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through down-Regulating MHC-Ι. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Masselli, E.; Vaccarezza, M.; Carubbi, C.; Pozzi, G.; Presta, V.; Mirandola, P.; Vitale, M. NK Cells: A Double Edge Sword against SARS-CoV-2. Adv. Biol. Regul. 2020, 77, 100737. [Google Scholar] [CrossRef]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and Nucleocapsid Proteins of SARS-CoV-2 Inhibit Type I Interferon Signaling Pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Silva Andrade, B.; Siqueira, S.; de Assis Soares, W.R.; de Souza Rangel, F.; Santos, N.O.; dos Santos Freitas, A.; Ribeiro da Silveira, P.; Tiwari, S.; Alzahrani, K.J.; Góes-Neto, A.; et al. Long-COVID and Post-COVID Health Complications: An Up-to-Date Review on Clinical Conditions and Their Possible Molecular Mechanisms. Viruses 2021, 13, 700. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in Older Adults: What We May Expect Regarding Pathogenesis, Immune Responses, and Outcomes. GeroScience 2020, 42, 505–514. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Xiao, N.; Nie, M.; Pang, H.; Wang, B.; Hu, J.; Meng, X.; Li, K.; Ran, X.; Long, Q.; Deng, H.; et al. Integrated Cytokine and Metabolite Analysis Reveals Immunometabolic Reprogramming in COVID-19 Patients with Therapeutic Implications. Nat. Commun. 2021, 12, 1618. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Merad, M.; Blish, C.A.; Sallusto, F.; Iwasaki, A. The Immunology and Immunopathology of COVID-19. Science 2022, 375, 1122–1127. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deitchman, A.N.; Torres, L.; Iyer, N.S.; Munter, S.E.; Nixon, C.C.; Donatelli, J.; Thanh, C.; Takahashi, S.; Hakim, J.; et al. Long-Term SARS-CoV-2-Specific Immune and Inflammatory Responses in Individuals Recovering from COVID-19 with and without Post-Acute Symptoms. Cell Rep. 2021, 36, 109518. [Google Scholar] [CrossRef]
- Wiech, M.; Chroscicki, P.; Swatler, J.; Stepnik, D.; De Biasi, S.; Hampel, M.; Brewinska-Olchowik, M.; Maliszewska, A.; Sklinda, K.; Durlik, M.; et al. Remodeling of T Cell Dynamics During Long COVID Is Dependent on Severity of SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 886431. [Google Scholar] [CrossRef]
- Maghool, F.; Valiani, A.; Safari, T.; Emami, M.H.; Mohammadzadeh, S. Gastrointestinal and Renal Complications in SARS-CoV-2-infected Patients: Role of Immune System. Scand. J. Immunol. 2021, 93. [Google Scholar] [CrossRef]
- de Oliveira, P.; Cunha, K.; Neves, P.; Muniz, M.; Gatto, G.; Salgado Filho, N.; Guedes, F.; Silva, G. Renal Morphology in Coronavirus Disease: A Literature Review. Med. Kaunas Lith. 2021, 57, 258. [Google Scholar] [CrossRef]
- Gabarre, P.; Dumas, G.; Dupont, T.; Darmon, M.; Azoulay, E.; Zafrani, L. Acute Kidney Injury in Critically Ill Patients with COVID-19. Intensive Care Med. 2020, 46, 1339–1348. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; De Greef, J.; Yildiz, H.; Pothen, L.; et al. SARS-CoV-2 Causes a Specific Dysfunction of the Kidney Proximal Tubule. Kidney Int. 2020, 98, 1296–1307. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. J. Am. Soc. Nephrol. JASN 2021, 32, 2851–2862. [Google Scholar] [CrossRef]
- Yende, S.; Parikh, C.R. Long COVID and Kidney Disease. Nat. Rev. Nephrol. 2021, 17, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Svetitsky, S.; Shuaib, R.; McAdoo, S.; Thomas, D.C. Long-Term Effects of COVID-19 on the Kidney. QJM Mon. J. Assoc. Physicians 2021, 114, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, E.; Hosseiniyan Khatibi, S.M.; Razi Soofiyani, S.; Abediazar, S.; Shoja, M.M.; Ardalan, M.; Zununi Vahed, S. COVID-19 and Kidney Injury: Pathophysiology and Molecular Mechanisms. Rev. Med. Virol. 2021, 31, e2176. [Google Scholar] [CrossRef] [PubMed]
- Carriazo, S.; Aparicio-Madre, M.I.; Tornero-Molina, F.; Fernández-Lucas, M.; Paraiso-Cuevas, V.; González-Parra, E.; Del Río-Gallegos, F.; Marques-Vidas, M.; Alcázar-Arroyo, R.; Martins-Muñoz, J.; et al. Impact of Different COVID-19 Waves on Kidney Replacement Therapy Epidemiology and Mortality: REMER 2020. Nephrol. Dial. Transplant. 2022, 37, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Basic-Jukic, N.; Racki, S.; Tolj, I.; Aleckovic, M.; Babovic, B.; Juric, I.; Furic-Cunko, V.; Katalinic, L.; Mihaljevic, D.; Vujic, S.; et al. Hospitalization and Death after Recovery from Acute COVID-19 among Renal Transplant Recipients. Clin. Transplant. 2022, 36, e14572. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.-G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 Promote SARS-CoV-2 Infection of Human Small Intestinal Enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Vodnar, D.-C.; Mitrea, L.; Teleky, B.-E.; Szabo, K.; Călinoiu, L.-F.; Nemeş, S.-A.; Martău, G.-A. Coronavirus Disease (COVID-19) Caused by (SARS-CoV-2) Infections: A Real Challenge for Human Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 575559. [Google Scholar] [CrossRef]
- Bogariu, A.M.; Dumitrascu, D.L. Digestive Involvement in the Long-COVID Syndrome. Med. Pharm. Rep. 2022, 95, 5–10. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Li, J.; Shen, L.; Zhu, L.; Liang, Y.; Lin, X.; Jiao, N.; Cheng, S.; Huang, Y.; et al. Gastrointestinal Sequelae 90 Days after Discharge for COVID-19. Lancet Gastroenterol. Hepatol. 2021, 6, 344–346. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of Antibody Immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and Predictors of Long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Raman, B.; Bluemke, D.A.; Lüscher, T.F.; Neubauer, S. Long COVID: Post-Acute Sequelae of COVID-19 with a Cardiovascular Focus. Eur. Heart J. 2022, 43, 1157–1172. [Google Scholar] [CrossRef]
- Bechmann, N.; Barthel, A.; Schedl, A.; Herzig, S.; Varga, Z.; Gebhard, C.; Mayr, M.; Hantel, C.; Beuschlein, F.; Wolfrum, C.; et al. Sexual Dimorphism in COVID-19: Potential Clinical and Public Health Implications. Lancet Diabetes Endocrinol. 2022, 10, 221–230. [Google Scholar] [CrossRef]
- Laha, S.; Chakraborty, J.; Das, S.; Manna, S.K.; Biswas, S.; Chatterjee, R. Characterizations of SARS-CoV-2 Mutational Profile, Spike Protein Stability and Viral Transmission. Infect. Genet. Evol. 2020, 85, 104445. [Google Scholar] [CrossRef]
- Antonelli, M.; Pujol, J.C.; Spector, T.D.; Ourselin, S.; Steves, C.J. Risk of Long COVID Associated with Delta versus Omicron Variants of SARS-CoV-2. The Lancet 2022, 399, 2263–2264. [Google Scholar] [CrossRef]
- Niemi, M.E.K.; Daly, M.J.; Ganna, A. The Human Genetic Epidemiology of COVID-19. Nat. Rev. Genet. 2022, 23, 533–546. [Google Scholar] [CrossRef]
- Caron, P. Thyroid Disorders and SARS-CoV-2 Infection: From Pathophysiological Mechanism to Patient Management. Ann. Endocrinol. 2020, 81, 507–510. [Google Scholar] [CrossRef]
- Gentile, S.; Strollo, F.; Mambro, A.; Ceriello, A. COVID -19, Ketoacidosis and New-onset Diabetes: Are There Possible Cause and Effect Relationships among Them? Diabetes Obes. Metab. 2020, 22, 2507–2508. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long COVID—Mechanisms, Risk Factors, and Management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Organelle | Cytopathic Manifestation(s) | Affected Host Protein(s) | Responsible SARS-CoV-2 Protein(s) | References |
|---|---|---|---|---|
| Mitochondria | Alteration of IFN-I responses | TOM70 | ORF9b protein | [39] |
| Dysregulating host antioxidant defense | SIRT1 | Nsp14 protein | [33,41] | |
| Endoplasmic reticulum | ER stress response, inhibition of IFN-β | IRE1 | ORF8, S, E, M proteins | [45,74] |
| Golgi Apparatus | Golgi fragmentation | GRASP55, TGN46 | S, M, E, nsp15, ORF3a proteins | [66,68] |
| Cytoskeleton | Cytoskeleton remodeling | Proteins of the MTOC | S protein | [66,89] |
| Ciliary dysfunction | CUL2 complex | ORF10 protein | [101] | |
| Cell membrane | Inhibition of immune synapse | T cell receptor | S protein | [106] |
| Nucleus | Inhibition of transcription factor entry into the nucleus | Transcription factors (such as STAT) | ORF6, ORF3b proteins | [116] |
| Inhibition of the transcription of IFN-stimulated genes | STAT, IRF3 | ORF6, ORF3b, N, nsp12 proteins | [114,116] | |
| Inhibition of host mRNA export from the nucleus | NXF1 | nsp1 protein | [119] |
| Organelle | Affected Cell Subset(s) or Regions | Cytopathy Manifestation(s) | References |
|---|---|---|---|
| Central nervous system | Cortex, hippocampus, and hypothalamus | Neuronal apoptosis through Caspase-3 | [128] |
| Ventral brain blood vessels | Structural alterations | [128,129] | |
| Microglia | Morphological alterations | [128] | |
| Limbic system | Microstructural changes and volume loss | [130] | |
| Respiratory system | Airway epithelium (ciliated cells, secretory cells, and type II alveolar cells) | Formation of viral plaques, cilia shedding and internalization, detachment of infected cells, cell death, and epithelial barrier damage | [114,131,132,133] |
| Multiciliated cells of the upper respiratory epithelium | Dedifferentiation through FOXJ1 downregulation | [134,135] | |
| Lower respiratory tract | Hyaline membrane or platelet-fibrin thrombus formation, accumulation of protein exudates, pneumocytic desquamation, diffuse alveolar damage, atypical pneumocytic hyperplasia, and syncytia | [122,131,136] | |
| Type II pneumocytes | Morphological alterations of mitochondria, accumulation of lipid and protein droplets, distended ER cisternae | [122,136] | |
| Circulatory system | Endothelium | Loss of endothelial integrity, cell detachment, endothelial dysfunction, alteration of the RAAS system, increased endothelial permeability, hypercoagulation, hypofibrinolysis, and prothrombotic states, increased mitochondrial ROS production, downregulation of ACE2 and eNOS | [24,137,138,139,140] |
| Inhibition of host mRNA export from the nucleus | nsp1 protein | [120] | |
| Cardiac tissue (especially cardiomyocytes) | Myocardial hypertrophy, cytokine production, acute myocardial infarction, interstitial edema, contractile deficits and sarcomere disassembly, necrosis, and cell death, lymphocytic inflammation, small vessel coronary artery disease | [141,142] | |
| Immune system | T cells | Severe lymphopenia, cytokine release syndrome, decreased IFN-γ-producing CD4+ T cell population, T cell depletion, alterations in Treg cells, T cell exhaustion | [81,143,144,145] |
| Lymph nodes and spleen | Necrosis, tissue degeneration, macrophage proliferation, atrophy | [143] | |
| Kidney | Renal cells (in general) | Cell detachment, tubular necrosis, inflammation of the ER, alterations of parietal epithelial cells, arteriosclerosis, protein absorption droplets, syncytia formation | [146] |
| Podocytes and proximal tubular cells | Hyperplasia, hypertrophy, tubular necrosis, loss of proteins in Bowman’s capsule, alteration of mitochondria, collapsing glomerulopathy | [146,147] | |
| Digestive system | Intestinal epithelium | Disruption of the epithelium integrity, syncytia formation, digestive tract motility and intestinal permeability, alteration of the intestinal microbiome and of the immune system, impaired function of mature enterocytes, overexpression of enzymes | [24,177,185] |
| Liver | Inflammation, hepatomegaly, hepatocytolysis, enzyme release | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Garcia, P.; Fiorillo Moreno, O.; Zarate Peñata, E.; Calderon-Villalba, A.; Pacheco Lugo, L.; Acosta Hoyos, A.; Villarreal Camacho, J.L.; Navarro Quiroz, R.; Pacheco Londoño, L.; Aroca Martinez, G.; et al. From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID. Int. J. Mol. Sci. 2023, 24, 8290. https://doi.org/10.3390/ijms24098290
Gonzalez-Garcia P, Fiorillo Moreno O, Zarate Peñata E, Calderon-Villalba A, Pacheco Lugo L, Acosta Hoyos A, Villarreal Camacho JL, Navarro Quiroz R, Pacheco Londoño L, Aroca Martinez G, et al. From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID. International Journal of Molecular Sciences. 2023; 24(9):8290. https://doi.org/10.3390/ijms24098290
Chicago/Turabian StyleGonzalez-Garcia, Pablo, Ornella Fiorillo Moreno, Eloina Zarate Peñata, Alejandro Calderon-Villalba, Lisandro Pacheco Lugo, Antonio Acosta Hoyos, Jose Luis Villarreal Camacho, Roberto Navarro Quiroz, Leonardo Pacheco Londoño, Gustavo Aroca Martinez, and et al. 2023. "From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID" International Journal of Molecular Sciences 24, no. 9: 8290. https://doi.org/10.3390/ijms24098290
APA StyleGonzalez-Garcia, P., Fiorillo Moreno, O., Zarate Peñata, E., Calderon-Villalba, A., Pacheco Lugo, L., Acosta Hoyos, A., Villarreal Camacho, J. L., Navarro Quiroz, R., Pacheco Londoño, L., Aroca Martinez, G., Moares, N., Gabucio, A., Fernandez-Ponce, C., Garcia-Cozar, F., & Navarro Quiroz, E. (2023). From Cell to Symptoms: The Role of SARS-CoV-2 Cytopathic Effects in the Pathogenesis of COVID-19 and Long COVID. International Journal of Molecular Sciences, 24(9), 8290. https://doi.org/10.3390/ijms24098290