
Spin Dynamics of Flavoproteins

 , and
, and
Abstract
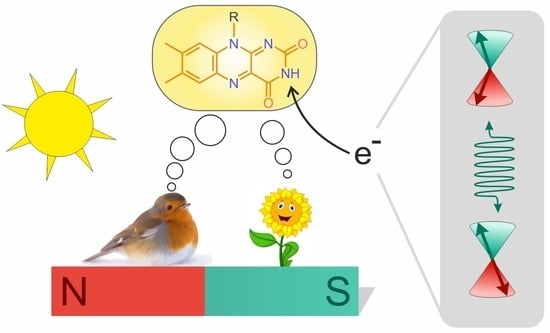
{kind=link}
{kind=link}
{kind=link}
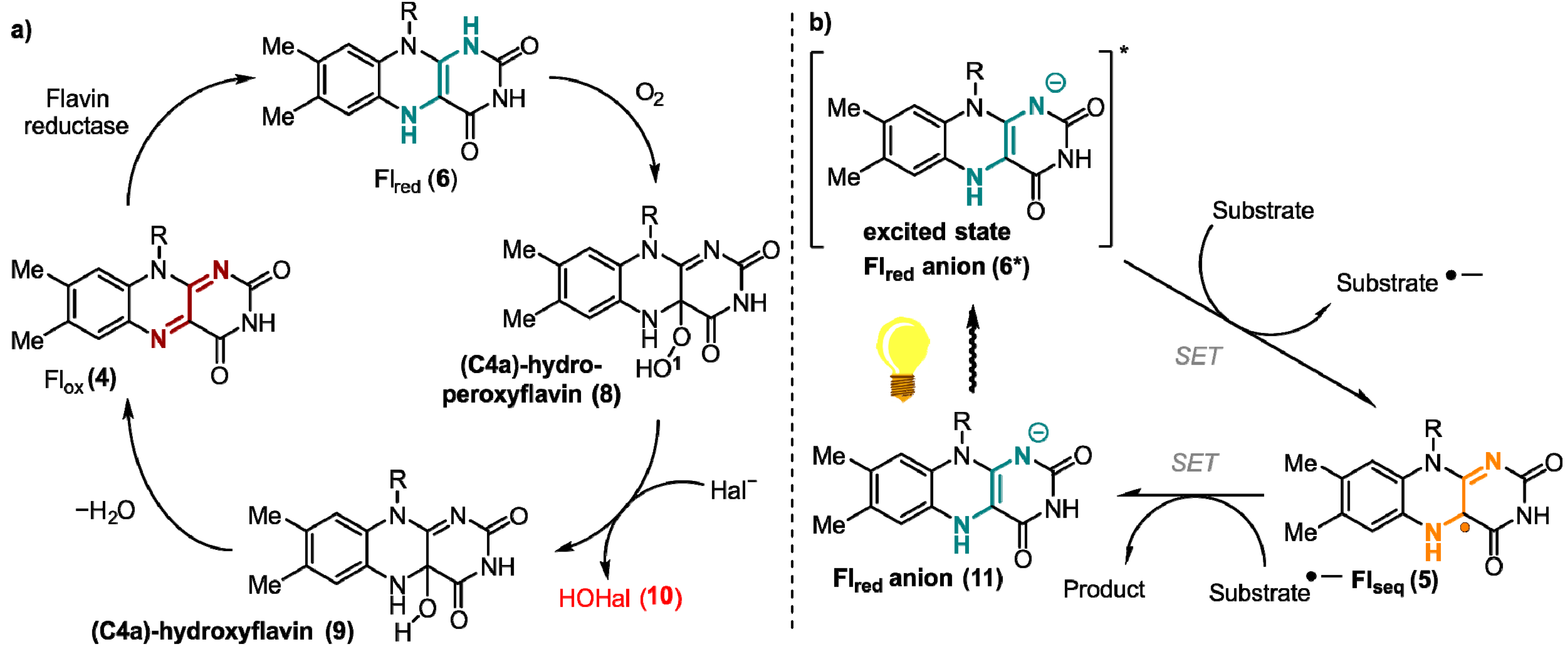{kind=link}
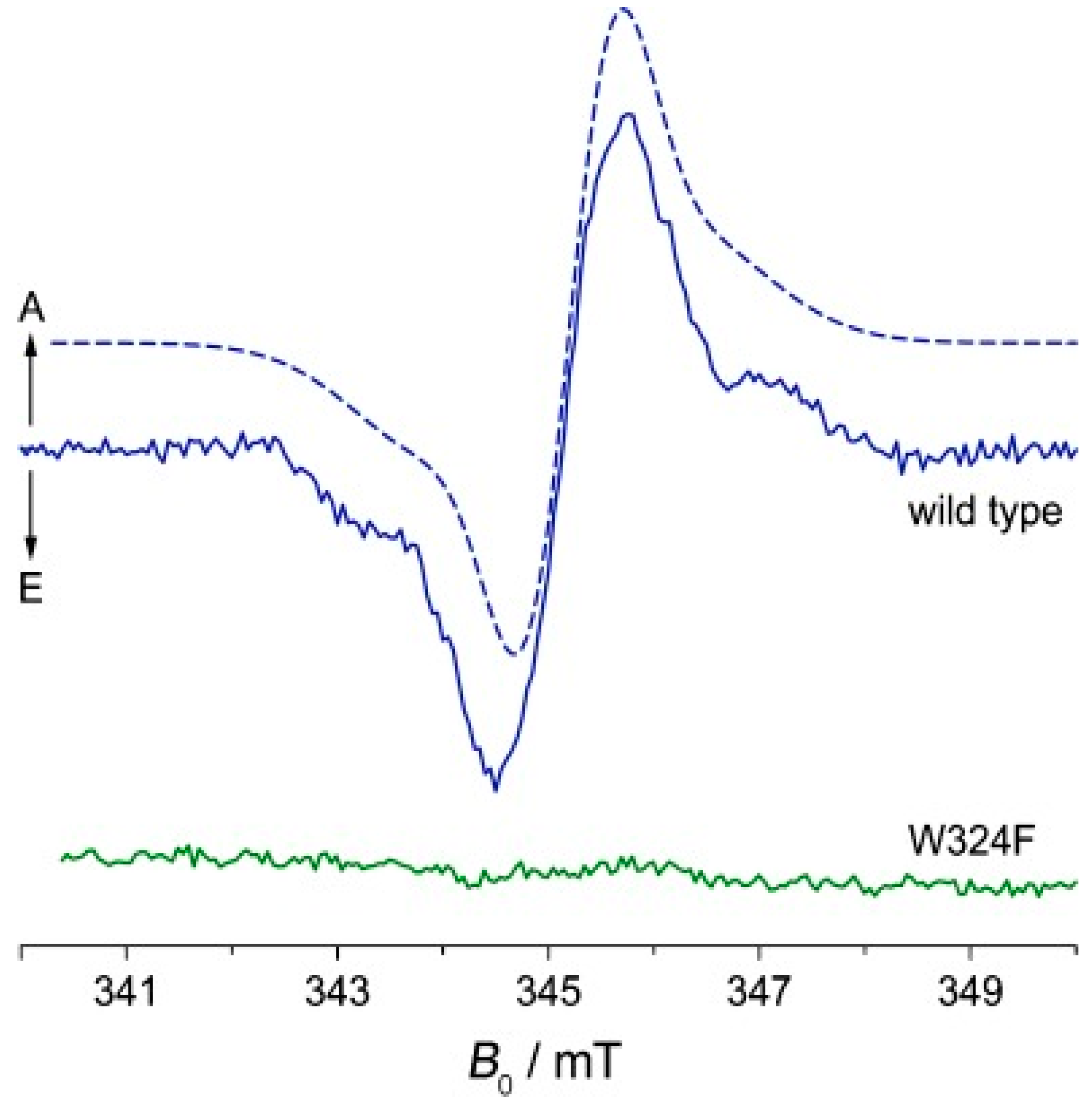{kind=link}
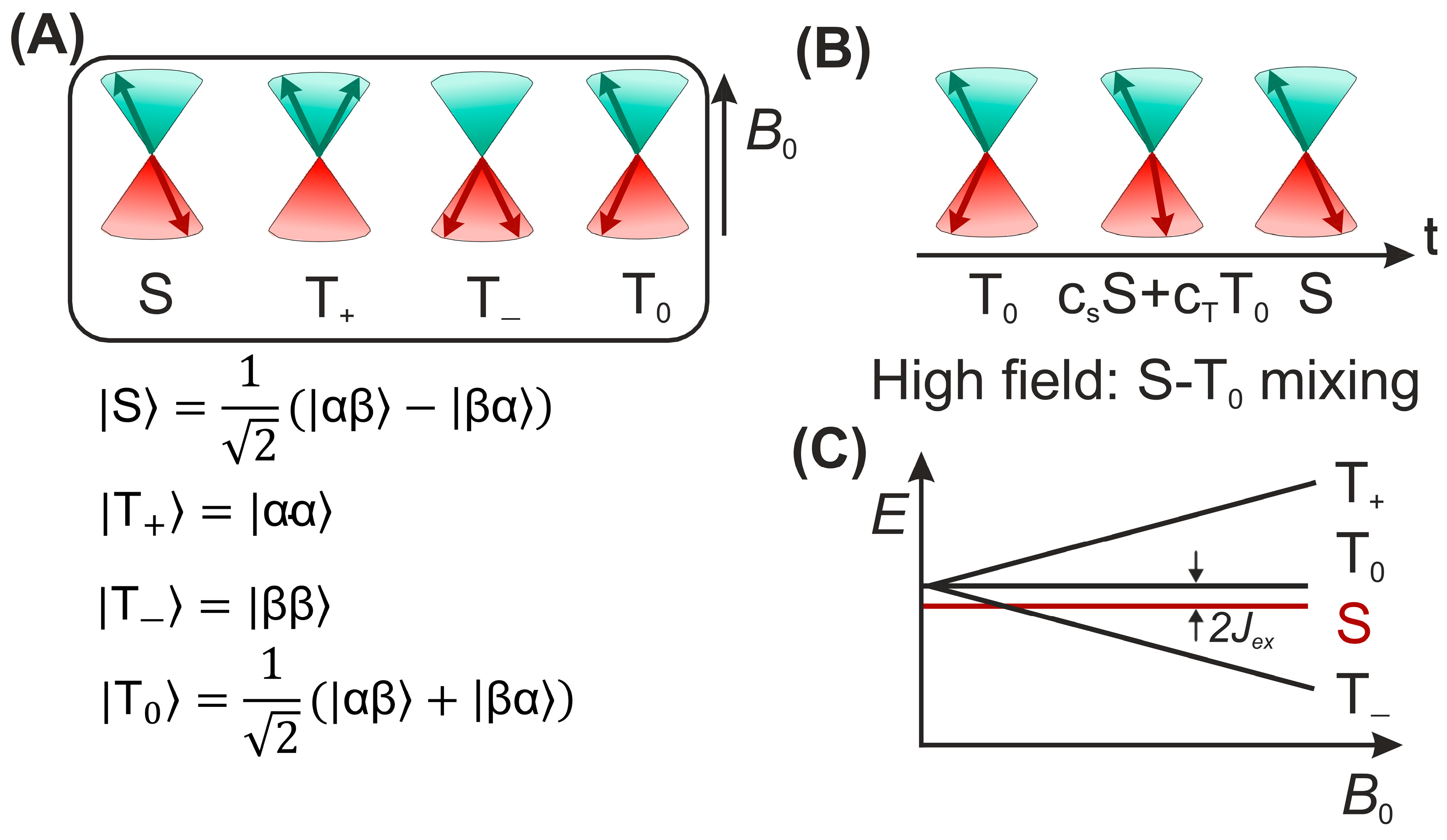{kind=link}
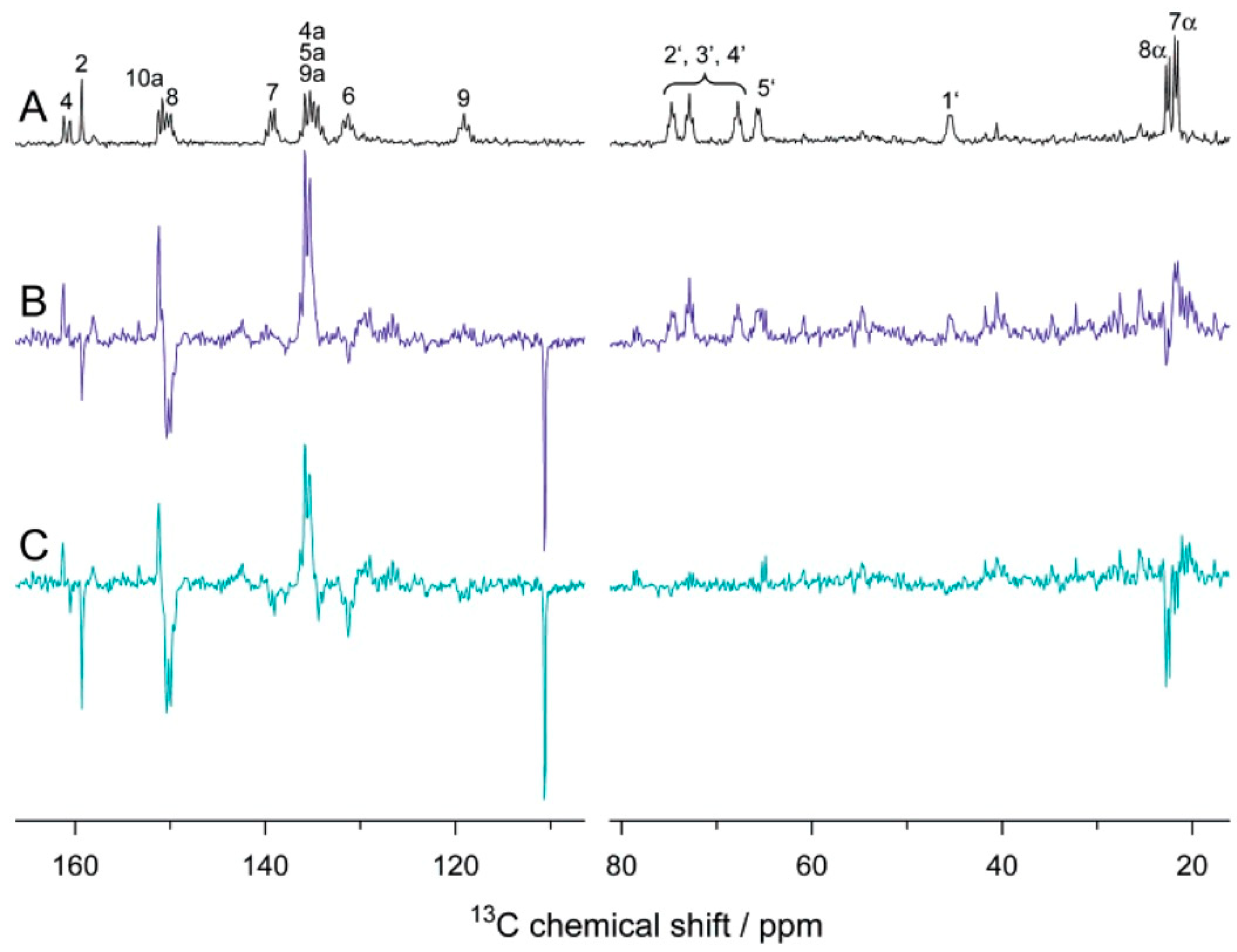{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
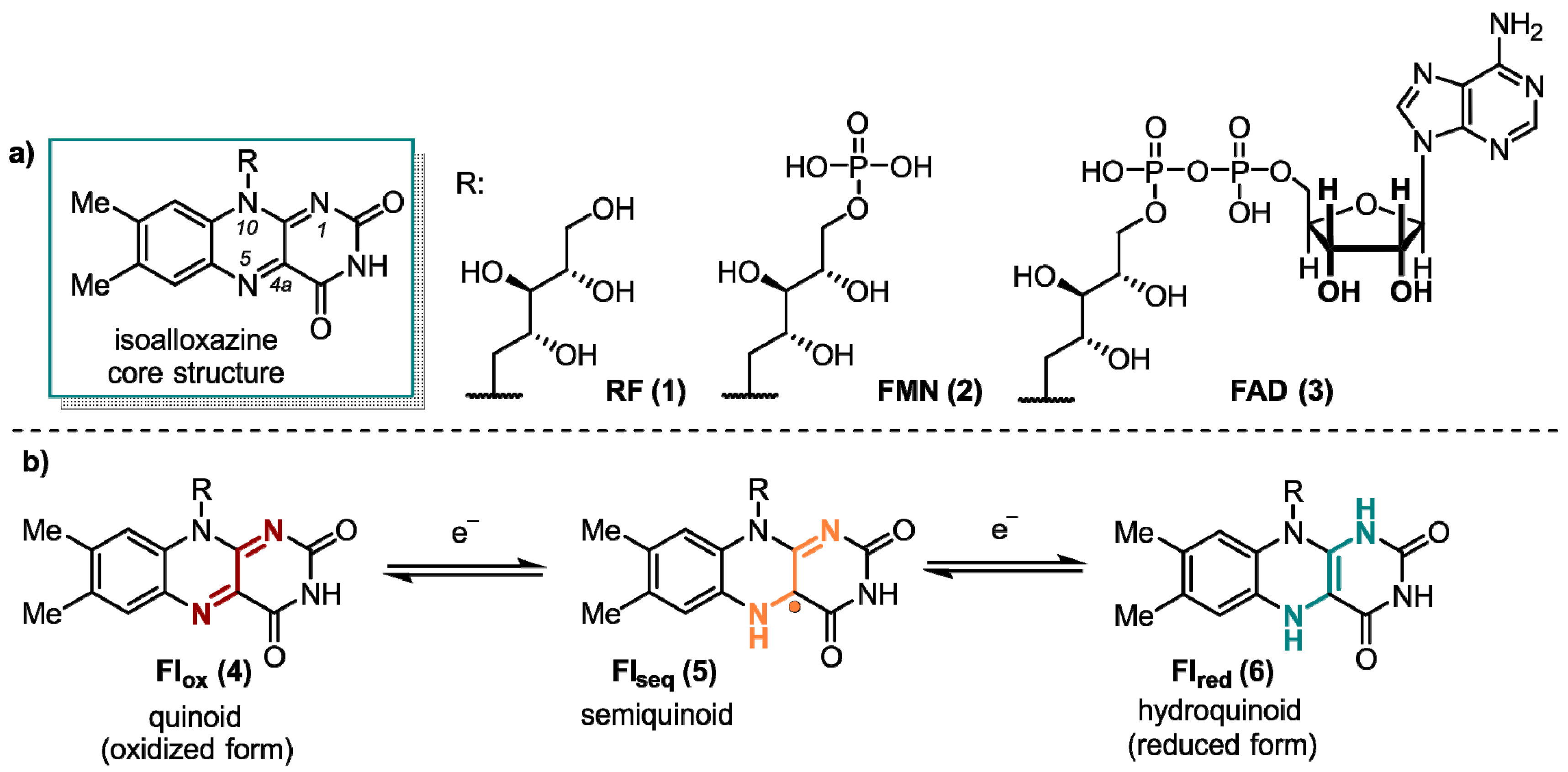
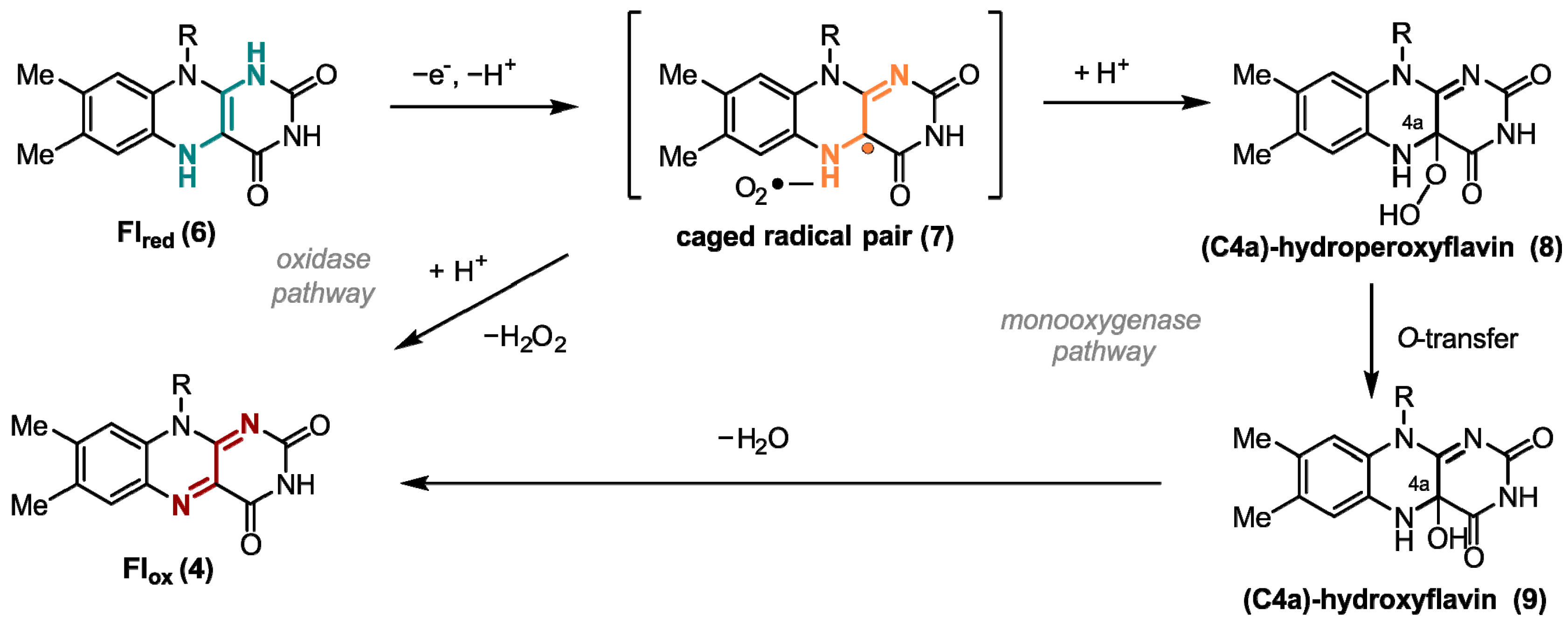
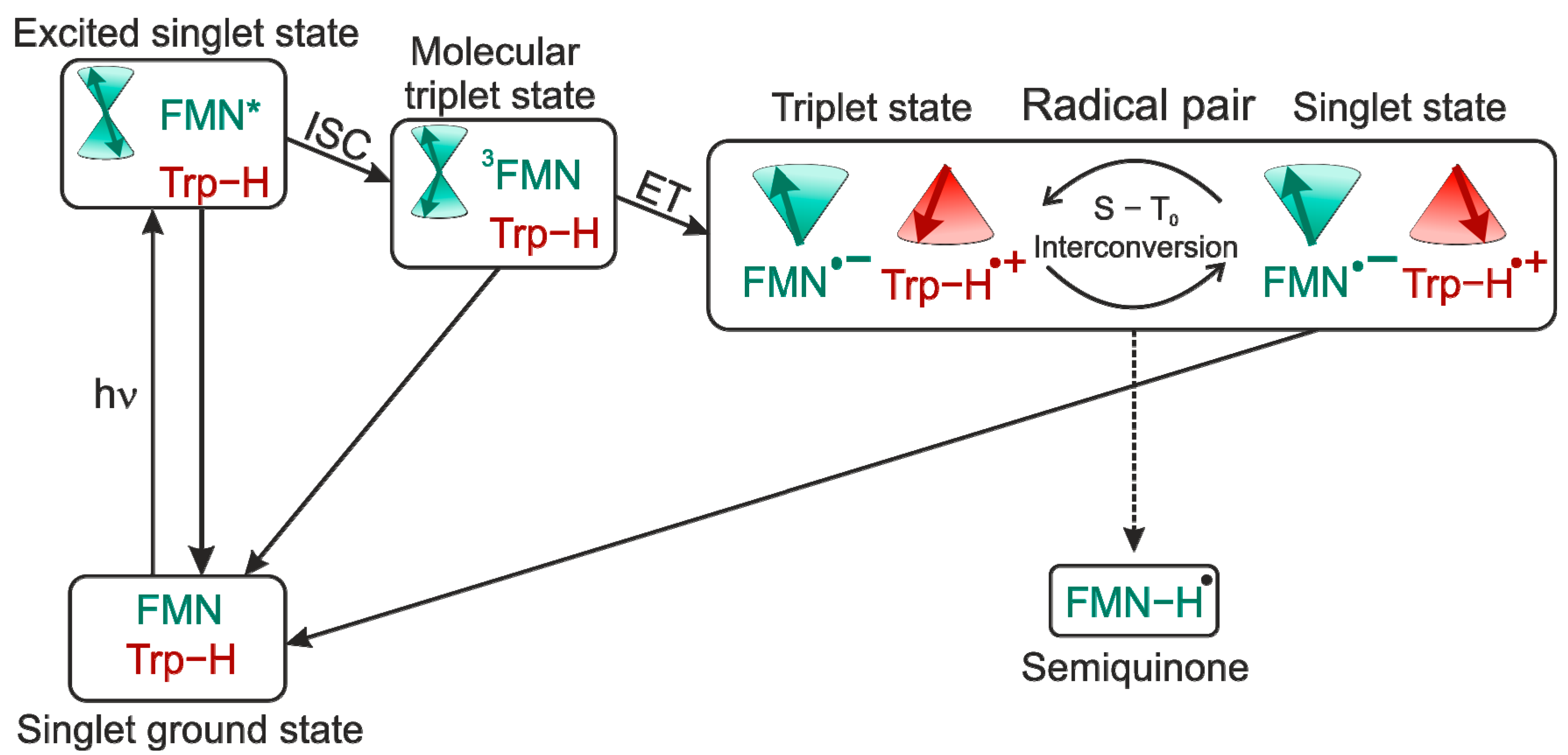
1. Flavins: Electronic Properties, Reactivity, and Photochemistry
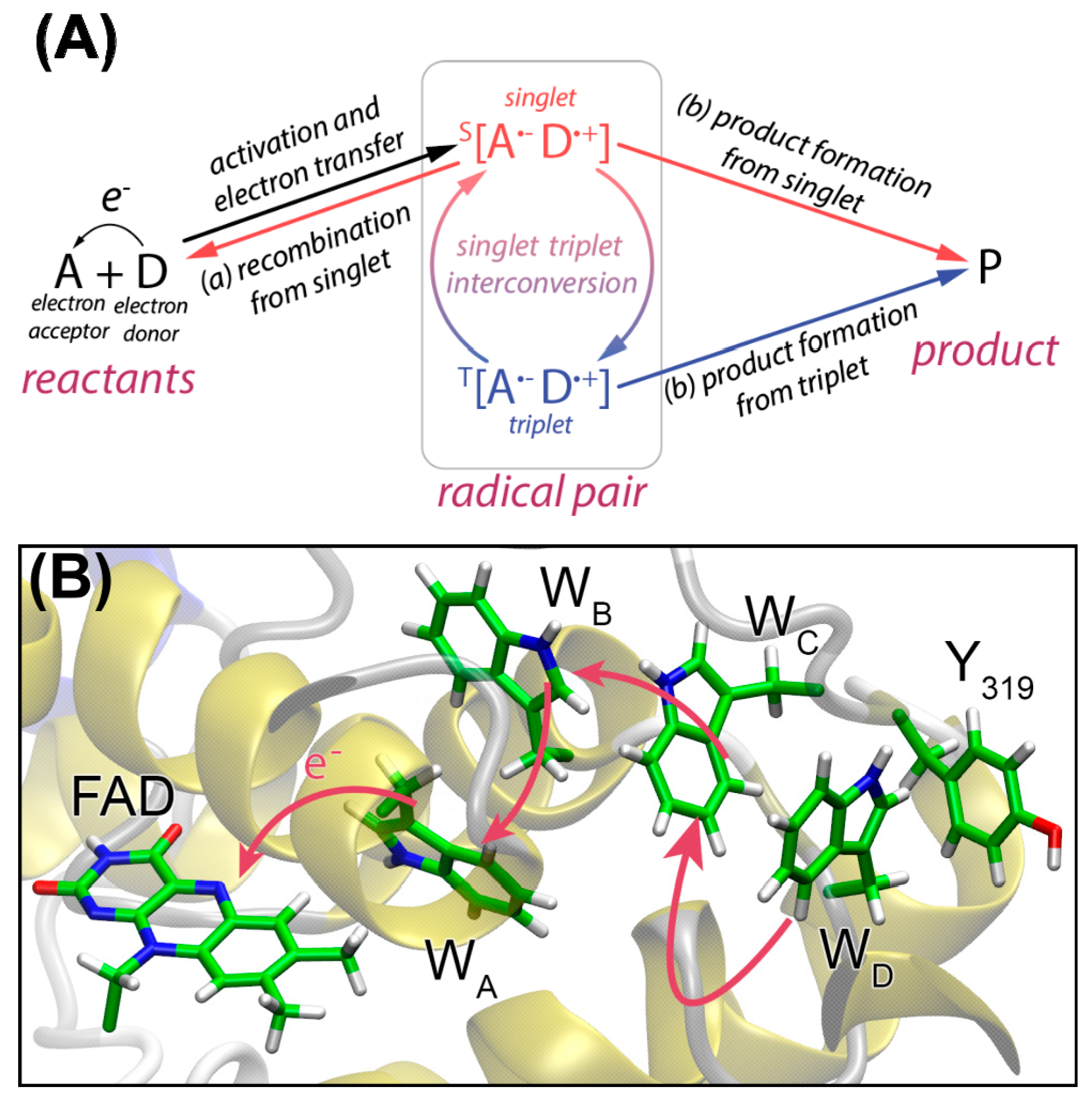
2. Flavins Acting in the Classical Radical Pair Mechanism in Liquid-State NMR
2.1. Discovery of the Principle of Photo-CIDNP in Liquid-State NMR
2.2. Flavoproteins Studied by Liquid-State Photo-CIDNP NMR
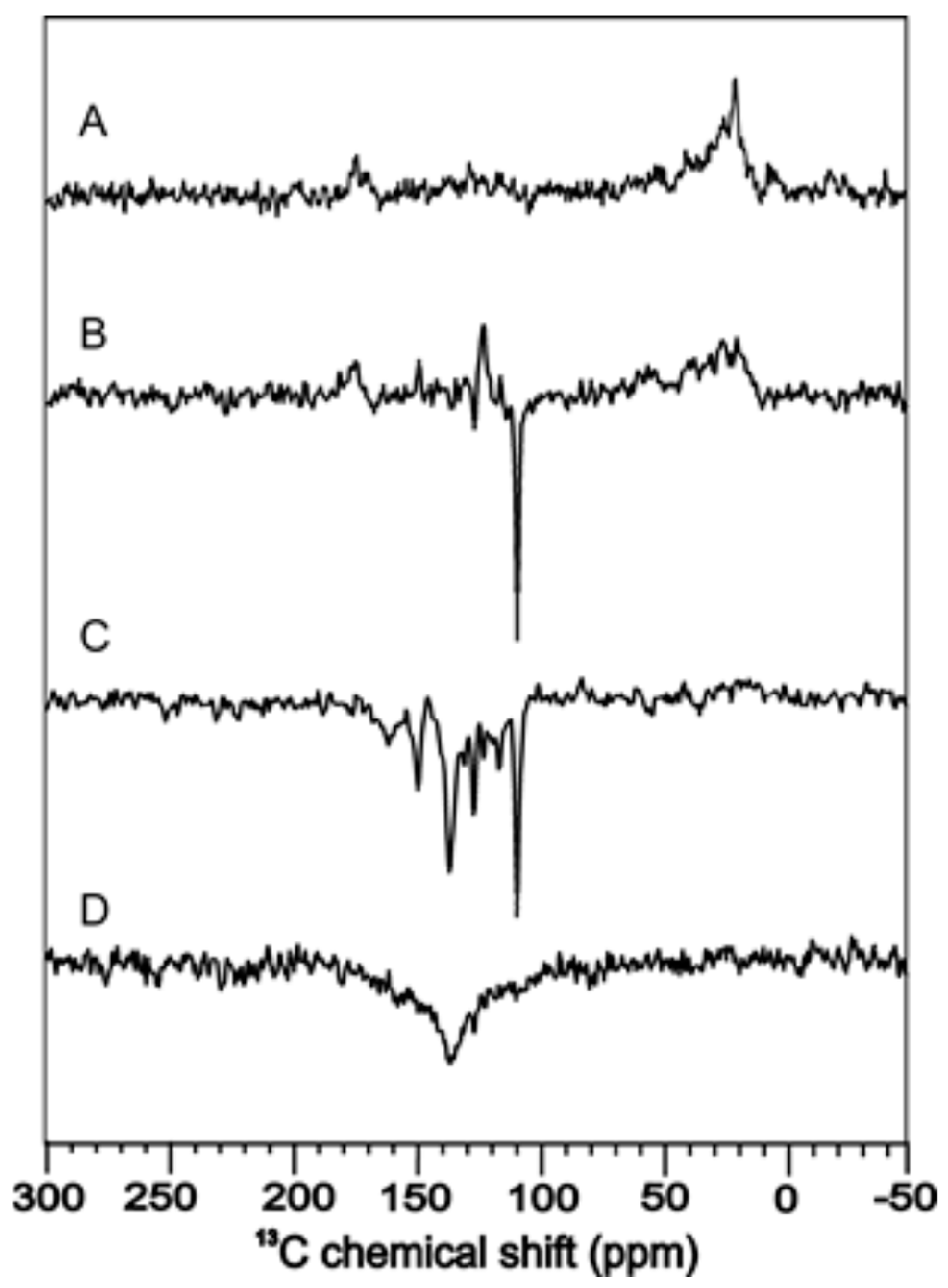
3. Flavoproteins Showing a Solid-State Photo-CIDNP Effect
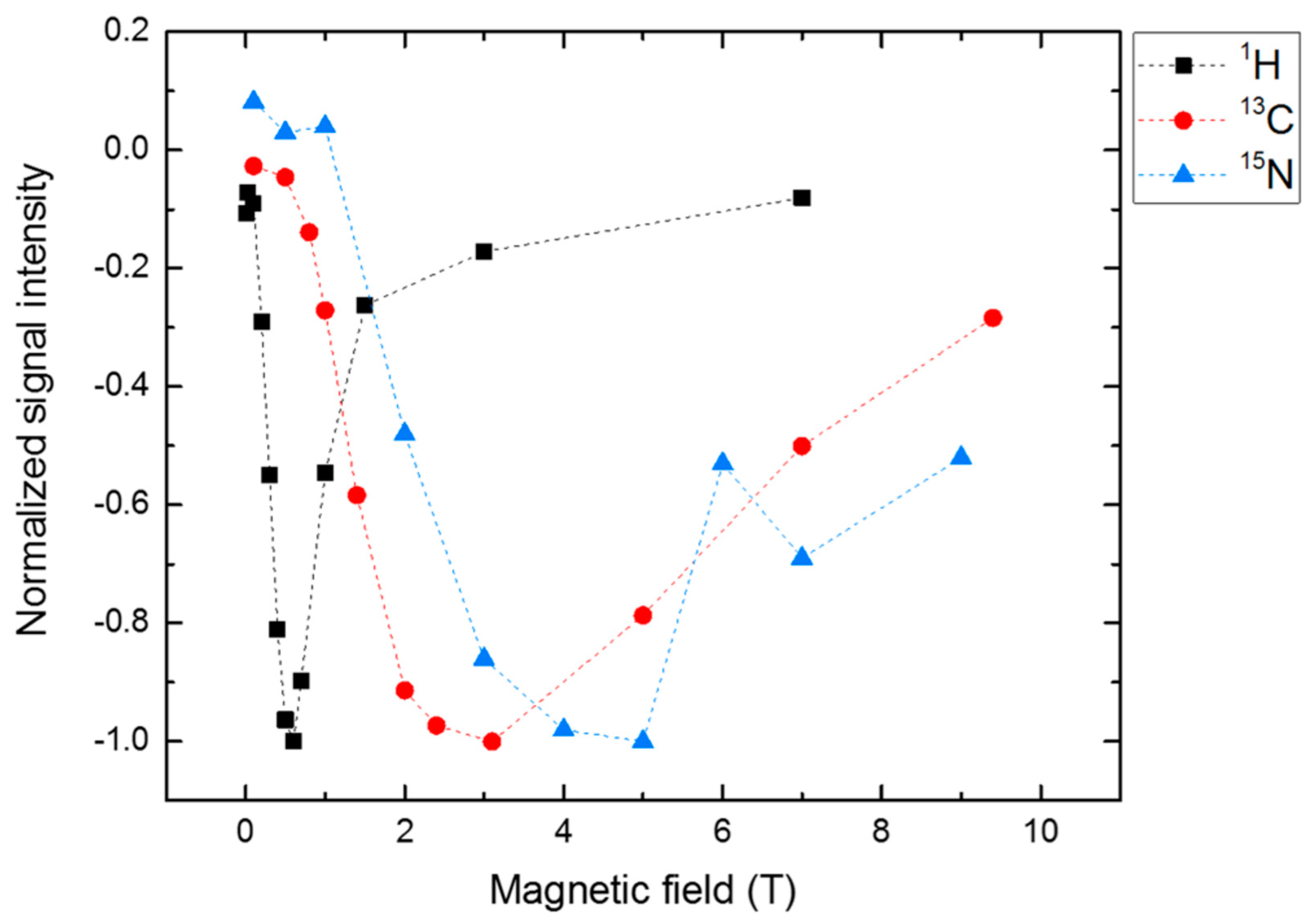
4. Spin-Dynamics of Flavoproteins in Animal Navigation
5. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silva, E.; Edwards, A.M. Flavins: Photochemistry and Photobiology; Royal Society of Chemistry: London, UK, 2006; ISBN 978-0-85404-331-6. [Google Scholar]
- Powers, H.J. Riboflavin (Vitamin B-2) and Health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Mansoorabadi, S.O.; Thibodeaux, C.J.; Liu, H.W. The Diverse Roles of Flavin Coenzymes—Nature’s Most Versatile Thespians. J. Org. Chem. 2007, 72, 6329–6342. [Google Scholar] [CrossRef] [PubMed]
- Manstein, D.J.; Pai, E.F. Purification and Characterization of FAD Synthetase from Brevibacterium Ammoniagenes. J. Biol. Chem. 1986, 261, 16169–16173. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Zhou, Q.; Mseeh, F.; Grishin, N.V.; Osterman, A.L.; Zhang, H. Crystal Structure of Human Riboflavin Kinase Reveals a β Barrel Fold and a Novel Active Site Arch. Structure 2003, 11, 265–273. [Google Scholar] [CrossRef]
- Northrop-Clewes, C.A.; Thurnham, D.I. The Discovery and Characterization of Riboflavin. Ann. Nutr. Metab. 2012, 61, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Yellow Enzyme and Its Functions. Biochem. Z. 1933, 266, 377–411. [Google Scholar]
- Krebs, H.A. Metabolism of Amino-Acids: Deamination of Amino-Acids. Biochem. J. 1935, 29, 1620–1644. [Google Scholar] [CrossRef]
- Theorell, H. Purification of the Active Group of the Yellow Enzyme. Biochem. Z. 1935, 275, 344–346. [Google Scholar]
- Tomiki, T.; Saitou, N. Phylogenetic Analysis of Proteins Associated in the Four Major Energy Metabolism Systems: Photosynthesis, Aerobic Respiration, Denitrification, and Sulfur Respiration. J. Mol. Evol. 2004, 59, 158–176. [Google Scholar] [CrossRef]
- Walsh, J.D.; Miller, A.F. Flavin Reduction Potential Tuning by Substitution and Bending. J. Mol. Struct. 2003, 623, 185–195. [Google Scholar] [CrossRef]
- Fraaijet, M.W.; Van Den Heuvel, R.H.H.; Van Berkel, W.J.H.; Mattevi, A. Covalent Flavinylation Is Essential for Efficient Redox Catalysis in Vanillyl-Alcohol Oxidase. J. Biol. Chem. 1999, 274, 35514–35520. [Google Scholar] [CrossRef] [PubMed]
- Dym, O.; Eisenberg, D. Sequence-Structure Analysis of FAD-Containing Proteins. Protein Sci. 2001, 10, 1712–1728. [Google Scholar] [CrossRef]
- McDonald, C.A.; Fagan, R.L.; Collard, F.; Monnier, V.M.; Palfey, B.A. Oxygen Reactivity in Flavoenzymes: Context Matters. J. Am. Chem. Soc. 2011, 133, 16809–16811. [Google Scholar] [CrossRef] [PubMed]
- Mattevi, A. To Be or Not to Be an Oxidase: Challenging the Oxygen Reactivity of Flavoenzymes. Trends Biochem. Sci. 2006, 31, 276–283. [Google Scholar] [CrossRef]
- Bučko, M.; Gemeiner, P.; Schenkmayerová, A.; Krajčovič, T.; Rudroff, F.; Mihovilovič, M.D. Baeyer-Villiger Oxidations: Biotechnological Approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef]
- Teufel, R.; Miyanaga, A.; Michaudel, Q.; Stull, F.; Louie, G.; Noel, J.P.; Baran, P.S.; Palfey, B.; Moore, B.S. Flavin-Mediated Dual Oxidation Controls an Enzymatic Favorskii-Type Rearrangement. Nature 2013, 503, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Wongnate, T.; Surawatanawong, P.; Visitsatthawong, S.; Sucharitakul, J.; Scrutton, N.S.; Chaiyen, P. Proton-Coupled Electron Transfer and Adduct Configuration Are Important for C4a-Hydroperoxyflavin Formation and Stabilization in a Flavoenzyme. J. Am. Chem. Soc. 2013, 136, 241–253. [Google Scholar] [CrossRef]
- Malmström, B.G. Enzymology of Oxygen. Annu. Rev. Biochem. 1982, 51, 21–59. [Google Scholar] [CrossRef]
- Romero, E.; Gómez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef]
- Wang, R.; Thorpe, C. Reactivity of Medium-Chain Acyl-CoA Dehydrogenase toward Molecular Oxygen. Biochemistry 1991, 30, 7895–7901. [Google Scholar] [CrossRef]
- Traber, R.; Kramer, H.E.A.; Hemmerich, P. One and Two Electron Transfer Pathways in the Photoreduction of Flavin. Pure Appl. Chem. 1982, 54, 1651–1665. [Google Scholar] [CrossRef]
- Büchler, J.; Papadopoulou, A.; Buller, R. Recent Advances in Flavin-Dependent Halogenase Biocatalysis: Sourcing, Engineering, and Application. Catalysts 2019, 9, 1030. [Google Scholar] [CrossRef]
- Andorfer, M.C.; Lewis, J.C. Understanding and Improving the Activity of Flavin-Dependent Halogenases via Random and Targeted Mutagenesis. Annu. Rev. Biochem. 2018, 87, 159–185. [Google Scholar] [CrossRef]
- Barker, R.D.; Yu, Y.; De Maria, L.; Johannissen, L.O.; Scrutton, N.S. Mechanism of Action of Flavin-Dependent Halogenases. ACS Catal. 2022, 12, 15352–15360. [Google Scholar] [CrossRef]
- Van Pée, K.H.; Patallo, E.P. Flavin-Dependent Halogenases Involved in Secondary Metabolism in Bacteria. Appl. Microbiol. Biotechnol. 2006, 70, 631–641. [Google Scholar] [CrossRef]
- Phintha, A.; Prakinee, K.; Chaiyen, P. Structures, Mechanisms and Applications of Flavin-Dependent Halogenases. In The Enzymes; Chaiyen, P., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 47, pp. 327–364. ISBN 9780128201374. [Google Scholar]
- Ahmad, I.; Vaid, F.H.M. Chapter 2: Photochemistry of Flavins in Aqueous and Organic Solvents. In Flavins: Photochemistry and Photobiology; Silva, E., Edwards, A.M., Eds.; Royal Society of Chemistry: London, UK, 2006; pp. 13–40. [Google Scholar]
- Hartman, T.; Cibulka, R. Photocatalytic Systems with Flavinium Salts: From Photolyase Models to Synthetic Tool for Cyclobutane Ring Opening. Org. Lett. 2016, 18, 3710–3713. [Google Scholar] [CrossRef]
- Scannell, M.P.; Fenick, D.J.; Yeh, S.R.; Falvey, D.E. Model Studies of DNA Photorepair: Reduction Potentials of Thymine and Cytosine Cyclobutane Dimers Measured by Fluorescence Quenching. J. Am. Chem. Soc. 1997, 119, 1971–1977. [Google Scholar] [CrossRef]
- Foja, R.; Walter, A.; Jandl, C.; Thyrhaug, E.; Hauer, J.; Storch, G. Reduced Molecular Flavins as Single-Electron Reductants after Photoexcitation. J. Am. Chem. Soc. 2022, 144, 4721–4726. [Google Scholar] [CrossRef]
- Rehpenn, A.; Walter, A.; Storch, G. Molecular Editing of Flavins for Catalysis. Synthesis 2021, 53, 2583–2593. [Google Scholar] [CrossRef]
- Seel, C.J.; Gulder, T. Biocatalysis Fueled by Light: On the Versatile Combination of Photocatalysis and Enzymes. ChemBioChem 2019, 20, 1871–1897. [Google Scholar] [CrossRef]
- Murray, A.T.; Matton, P.; Fairhurst, N.W.G.; John, M.P.; Carbery, D.R. Biomimetic Flavin-Catalyzed Aldehyde Oxidation. Org. Lett. 2012, 14, 3656–3659. [Google Scholar] [CrossRef]
- Graml, A.; Neveselý, T.; Jan Kutta, R.; Cibulka, R.; König, B. Deazaflavin Reductive Photocatalysis Involves Excited Semiquinone Radicals. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mojr, V.; Svobodová, E.; Straková, K.; Neveselý, T.; Chudoba, J.; Dvořáková, H.; Cibulka, R. Tailoring Flavins for Visible Light Photocatalysis: Organocatalytic [2+2] Cycloadditions Mediated by a Flavin Derivative and Visible Light. Chem. Commun. 2015, 51, 12036–12039. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Storch, G. Synthetic C6-Functionalized Aminoflavin Catalysts Enable Aerobic Bromination of Oxidation-Prone Substrates. Angew. Chem. Int. Ed. 2020, 59, 22505–22509. [Google Scholar] [CrossRef]
- Xu, J.; Jarocha, L.E.; Zollitsch, T.; Konowalczyk, M.; Henbest, K.B.; Richert, S.; Golesworthy, M.J.; Schmidt, J.; Déjean, V.; Sowood, D.J.C.; et al. Magnetic Sensitivity of Cryptochrome 4 from a Migratory Songbird. Nature 2021, 594, 535–540. [Google Scholar] [CrossRef]
- Evans, E.W.; Dodson, C.A.; Maeda, K.; Biskup, T.; Wedge, C.J.; Timmel, C.R. Magnetic Field Effects in Flavoproteins and Related Systems. Interface Focus 2013, 3, 20130037. [Google Scholar] [CrossRef]
- Paul, S.; Meng, L.; Berger, S.; Grampp, G.; Matysik, J.; Wang, X. The Flavin–Tryptophan Dyad F10T as a Cryptochrome Model Compound: Synthesis and Photochemistry. ChemPhotoChem 2017, 1, 12–16. [Google Scholar] [CrossRef]
- Paul, S.; Kiryutin, A.S.; Guo, J.; Ivanov, K.L.; Matysik, J.; Yurkovskaya, A.V.; Wang, X. Magnetic Field Effect in Natural Cryptochrome Explored with Model Compound. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Hore, P.J.; Mouritsen, H. The Radical-Pair Mechanism of Magnetoreception. Annu. Rev. Biophys. 2016, 45, 299–344. [Google Scholar] [CrossRef]
- Messiha, H.L.; Wongnate, T.; Chaiyen, P.; Jones, A.R.; Scrutton, N.S. Magnetic Field Effects as a Result of the Radical Pair Mechanism Are Unlikely in Redox Enzymes. J. R. Soc. Interface 2015, 12, 20141155. [Google Scholar] [CrossRef]
- Bargon, J.; Fischer, H.; Johnsen, U. Kernresonanz-Emissionslinien Während Rascher Radikalreaktionen I. Aufnahmeverfahren Und Beispiele. Z. Naturforsch. A 1967, 22, 1551–1555. [Google Scholar] [CrossRef]
- Ward, F.R.; Lawler, R.G. Nuclear Magnetic Resonance Emission and Enhanced Absorption in Rapid Organometallic Reactions. J. Am. Chem. Soc. 1967, 89, 5518–5519. [Google Scholar] [CrossRef]
- Cocivera, M. Optically Induced Overhauser Effect in Solution. Nuclear Magnetic Resonance Emission. J. Am. Chem. Soc. 1968, 90, 3261–3263. [Google Scholar] [CrossRef]
- Fessenden, R.W.; Schuler, R.H. Electron Spin Resonance Studies of Transient Alkyl Radicals. J. Chem. Phys. 1963, 39, 2147–2195. [Google Scholar] [CrossRef]
- Smaller, B.; Remko, J.R.; Avery, E.C. Electron Paramagnetic Resonance Studies of Transient Free Radicals Produced by Pulse Radiolysis. J. Chem. Phys. 1968, 48, 5174–5181. [Google Scholar] [CrossRef]
- Paul, H.; Fischer, H. Emission Und Verstärkte Absorption in Den ESR-Spektren Kurzlebiger Radikale in Wäßrigen Lösungen. Z. Naturforsch. A 1970, 25, 443–445. [Google Scholar] [CrossRef]
- Fischer, H.; Bargon, J. Chemically Induced Dynamic Nuclear Polarization during Thermal Decomposition of Peroxides and Azo Compounds. Acc. Chem. Res. 1969, 2, 110–114. [Google Scholar] [CrossRef]
- Bargon, J.; Fischer, H. Kernresonanz-Emissionslinien Während Rascher Radikalreaktionen II. Chemisch Induzierte Dynamische Kernpolarisation. Z. Naturforsch. A 1967, 22, 1556–1562. [Google Scholar] [CrossRef]
- Lawler, R.G. Chemically Induced Dynamic Nuclear Polarization. J. Am. Chem. Soc. 1967, 89, 5519–5521. [Google Scholar] [CrossRef]
- Kaptein, R. Chemically Induced Dynamic Nuclear Polarization in Five Alkyl Radicals. Chem. Phys. Lett. 1968, 2, 261–267. [Google Scholar] [CrossRef]
- Closs, G.L.; Closs, L.E. Induced Dynamic Nuclear Spin Polarization in Reactions of Photochemically and Thermally Generated Triplet Diphenylmethylene. J. Am. Chem. Soc. 1969, 91, 4549–4550. [Google Scholar] [CrossRef]
- Closs, G.L.; Trifunac, A.D. Chemically Induced Nuclear Spin Polarization as a Tool for Determination of Spin Multiplicities of Radical-Pair Precursors. J. Am. Chem. Soc. 1969, 91, 4554–4555. [Google Scholar] [CrossRef]
- Kaptein, R.; Oosterhoff, J.L. Chemically Induced Dynamic Nuclear Polarization II. (Relation with Anomalous ESR Spectra). Chem. Phys. Lett. 1969, 4, 195–197. [Google Scholar] [CrossRef]
- Closs, G.L.; Forbes, M.D.E.; Norris, J.R. Spin-Polarized Electron Paramagnetic Resonance Spectra of Radical Pairs in Micelles: Observation of Electron Spin-Spin Interactions. J. Phys. Chem. 1987, 91, 3592–3599. [Google Scholar] [CrossRef]
- Hore, P.J.; Hunter, D.A.; McKie, C.D.; Hoff, A.J. Electron Paramagnetic Resonance of Spin-Correlated Radical Pairs in Photosynthetic Reactions. Chem. Phys. Lett. 1987, 137, 495–500. [Google Scholar] [CrossRef]
- Biskup, T.; Schleicher, E.; Okafuji, A.; Link, G.; Hitomi, K.; Getzoff, E.D.; Weber, S. Direct Observation of a Photoinduced Radical Pair in a Cryptochrome Blue-Light Photoreceptor. Angew. Chem. Intl. Ed. 2009, 48, 404–407. [Google Scholar] [CrossRef]
- Matysik, J.; Ding, Y.; Kim, Y.; Kurle, P.; Yurkovskaya, A.; Ivanov, K.; Alia, A. Photo-CIDNP in Solid State. Appl. Magn. Reson. 2022, 53, 521–537. [Google Scholar] [CrossRef]
- Adrian, F.J. Principles of the Radical Pair Mechanism of Chemically Induced Nuclear and Electron Spin Polarization. Rev. Chem. Intermed. 1979, 3, 3–43. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Principles of Molecular Photochemistry: An Introduction; University Science Books: Sausalito, CA, USA, 2009; ISBN 978-1-891389-57-3. [Google Scholar]
- Panov, M.S.; Pravdivtsev, A.N.; Ivanov, K.L.; Yurkovskaya, A.V.; Vieth, H.-M. Coherent Polarization Transfer Effects Are Crucial for Interpreting Low-Field CIDNP Data. Appl. Magn. Reson. 2014, 45, 893–900. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Pravdivtsev, A.N.; Yurkovskaya, A.V.; Vieth, H.M.; Kaptein, R. The Role of Level Anti-Crossings in Nuclear Spin Hyperpolarization. Prog. Nuc. Magn. Reson. Spectrosc. 2014, 81, 1–36. [Google Scholar] [CrossRef]
- Salikhov, K.M.; Molin, Y.N.; Sagdeev, R.Z.; Buchachenko, A.L. Spin Polarization and Magnetic Effects in Radical Reactions; Elsiever: Amsterdam, The Netherlands, 1984; ISBN 0-444-99677-X. [Google Scholar]
- Kaptein, R. Simple Rules for Chemically Induced Dynamic Nuclear Polarization. J. Chem. Soc. D 1971, 732–733. [Google Scholar] [CrossRef]
- Kaptein, R. Chemically Induced Dynamic Nuclear Polarization. VIII. Spin Dynamics and Diffusion of Radical Pairs. J. Am. Chem. Soc. 1972, 94, 6251–6262. [Google Scholar] [CrossRef]
- Köckenberger, W.; Matysik, J. Hyperpolarization Methods in NMR; Topics in Current Chemistry; Kuhn, L.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 338, ISBN 9780128032244. [Google Scholar]
- Salikhov, K.M. Mutual Effect of Nuclei upon CIDNP in High Fields. Violation of the Kaptein Rules. Chem. Phys. 1982, 64, 371–379. [Google Scholar] [CrossRef]
- Hore, J.; Broadhurst, R.W. Photo-CIDNP of Biopolymers. Prog. Nucl. Magn. Reson. Spectrosc. 1993, 25, 345–402. [Google Scholar] [CrossRef]
- Obynochny, A.A.; Sagdeev, R.Z.; Salikhov, K.M.; Sukhenko, S.A.; Yurkovskaya, A.V. Optical Nuclear Polarization in the Gas Phase. Chem. Phys. 1986, 104, 415–419. [Google Scholar] [CrossRef]
- Yurkovskaya, A.V.; Galimov, R.R.; Obynochny, A.A.; Salikhov, K.M.; Sagdeev, R.Z. The Field Dependence of CIDNP in Gas-Phase Reactions of Biradicals. Chem. Phys. 1987, 112, 259–264. [Google Scholar] [CrossRef]
- Okuno, Y.; Cavagnero, S. Fluorescein: A Photo-CIDNP Sensitizer Enabling Hypersensitive NMR Data Collection in Liquids at Low Micromolar Concentration. J. Phys. Chem. B 2016, 120, 715–723. [Google Scholar] [CrossRef]
- Sadler, D.E.; Wendler, J.; Olbrich, G.; Schaffner, K. Photochemical Reaction Mechanisms of β,γ-Unsaturated Ketones. The 1,3-Acetyl Shift in Cyclopent-2-Enyl Methyl Ketones. J. Am. Chem. Soc. 1984, 106, 2064–2071. [Google Scholar] [CrossRef]
- Roth, H.D. Organic Radical Cations in Fluid Solution: Unusual Structures and Rearrangements. Acc. Chem. Res. 1987, 20, 343–350. [Google Scholar] [CrossRef]
- Neshchadin, D.; Gescheidt, G. CIDNP as a Tool for Rapid Rearrangements: New Insights into Cyclobutanoid 4-Center/3-Electron Radical Cations. J. Phys. Org. Chem. 2013, 26, 737–741. [Google Scholar] [CrossRef]
- Kaptein, R. Photo-CIDNP Studies of Proteins. In Biological Magnetic Resonance; Springer: Boston, MA, USA, 1982; pp. 145–191. [Google Scholar]
- Mok, K.H.; Hore, P.J. Photo-CIDNP NMR Methods for Studying Protein Folding. Methods 2004, 34, 75–87. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Miesel, K.; Yurkovskaya, A.V.; Korchak, S.E.; Kiryutin, A.S.; Vieth, H.-M. Transfer of CIDNP among Coupled Spins at Low Magnetic Field. Appl. Magn. Reson. 2006, 30, 513–534. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Yurkovskaya, A.V.; Vieth, H.-M. Coherent Transfer of Hyperpolarization in Coupled Spin Systems at Variable Magnetic Field. J. Chem. Phys. 2008, 128, 154701. [Google Scholar] [CrossRef] [PubMed]
- Pravdivtsev, A.N.; Yurkovskaya, A.V.; Ivanov, K.L.; Vieth, H.M. Importance of Polarization Transfer in Reaction Products for Interpreting and Analyzing CIDNP at Low Magnetic Fields. J. Magn. Reson. 2015, 254, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Yurkovskaya, A.; Grosse, S.; Dvinskikh, S.; Morozova, O.; Vieth, H.-M. Spin and Molecular Dynamics of Biradicals as Studied by Low Field Nuclear Polarization at Variable Temperature. J. Phys. Chem. A 1999, 103, 980–988. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Tarasov, V.F.; Bagryanskaya, E.G. Electron Spin Exchange in Micellized Radical Pairs. I. 13C Low Field Chemically Induced Dynamic Nuclear Polarization (CIDNP) and 13C Radio Frequency Stimulated Nuclear Polarization (SNP). Chem. Phys. 1991, 153, 427–441. [Google Scholar] [CrossRef]
- Lopez, J.J.; Carter, M.A.G.; Tsentalovich, Y.P.; Morozova, O.B.; Yurkovskaya, A.V.; Hore, P.J. Effects of Surfactants on the Photosensitized Production of Tyrosine Radicals Studied by Photo-CIDNP. Photochem. Photobiol. 2002, 75, 6. [Google Scholar] [CrossRef]
- Morozova, O.B.; Yurkovskaya, A.V.; Tsentalovich, Y.P.; Vieth, H.-M. 1H and 13C Nuclear Polarization in Consecutive Biradicals during the Photolysis of 2,2,12,12-Tetramethylcyclododecanone. J. Phys. Chem. A 1997, 101, 399–406. [Google Scholar] [CrossRef]
- Morozova, O.B.; Yurkovskaya, A.V.; Tsentalovich, Y.P.; Sagdeev, R.Z.; Wu, T.; Forbes, M.D.E. Study of Consecutive Biradicals from 2-Hydroxy-2,12-Dimethylcyclododecanone by TR-CIDNP, TREPR, and Laser Flash Photolysis. J. Phys. Chem. A 1997, 101, 8803–8808. [Google Scholar] [CrossRef]
- Tsentalovich, Y.P.; Morozova, O.B.; Avdievich, N.I.; Ananchenko, G.S.; Yurkovskaya, A.V.; Ball, J.D.; Forbes, M.D.E. Influence of Molecular Structure on the Rate of Intersystem Crossing in Flexible Biradicals. J. Phys. Chem. A 1997, 101, 8809–8816. [Google Scholar] [CrossRef]
- Müller, F.; Hemmerich, P.; Ehrenberg, A.; Palmer, G.; Massey, V. The Chemical and Electronic Structure of the Neutral Flavin Radical as Revealed by Electron Spin Resonance Spectroscopy of Chemically and Isotopically Substituted Derivatives. Eur. J. Biochem. 1970, 14, 185–196. [Google Scholar] [CrossRef]
- Edmondson, D.E. Electron-Spin-Resonance Studies on Flavoenzymes. Biochem. Soc. Trans. 1985, 13, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, A.; Müller, F.; Hemmerich, P. Basicity, Visible Spectra, and Electron Spin Resonance of Flavosemiquinone Anions. Eur. J. Biochem. 1967, 2, 286–293. [Google Scholar] [CrossRef]
- Bretz, N.; Mastalsky, I.; Elsner, M.; Kurreck, H. First ENDOR Investigations of Biologically Relevant Organic Radicals in Reversed Micelles. Angew. Chem. Int. Ed. 1987, 26, 345–347. [Google Scholar] [CrossRef]
- Massey, V. The Chemical and Biological Versatility of Riboflavin. Biochem. Soc. Trans. 2000, 28, 283. [Google Scholar] [CrossRef]
- Frey, P.A. Radical Mechanisms of Enzymatic Catalysis. Annu. Rev. Biochem. 2001, 70, 121–148. [Google Scholar] [CrossRef]
- Swartz, T.E.; Corchnoy, S.B.; Christie, J.M.; Lewis, J.W.; Szundi, I.; Briggs, W.R.; Bogomolni, R.A. The Photocycle of a Flavin-Binding Domain of the Blue Light Photoreceptor Phototropin. J. Biol. Chem 2001, 276, 36493–36500. [Google Scholar] [CrossRef]
- Kennis, J.T.M.; Crosson, S.; Gauden, M.; Van Stokkum, I.H.M.; Moffat, K.; Van Grondelle, R. Primary Reactions of the LOV2 Domain of Phototropin, a Plant Blue-Light Photoreceptor. Biochemistry 2003, 42, 3385–3392. [Google Scholar] [CrossRef]
- Salomon, M.; Christie, J.M.; Knieb, E.; Lempert, U.; Briggs, W.R. Photochemical and Mutational Analysis of the FMN-Binding Domains of the Plant Blue Light Receptor, Phototropin. Biochemistry 2000, 39, 9401–9410. [Google Scholar] [CrossRef]
- Richter, G.; Weber, S.; Römisch, W.; Bacher, A.; Fischer, M.; Eisenreich, W. Photochemically Induced Dynamic Nuclear Polarization in a C450A Mutant of the LOV2 Domain of the Avena Sativa Blue-Light Receptor Phototropin. J. Am. Chem. Soc. 2005, 127, 17245–17252. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.; Kowalczyk, R.M.; Kay, C.W.M.; Hegemann, P.; Bacher, A.; Fischer, M.; Bittl, R.; Richter, G.; Weber, S. On the Reaction Mechanism of Adduct Formation in LOV Domains of the Plant Blue-Light Receptor Phototropin. J. Am. Chem. Soc. 2004, 126, 11067–11076. [Google Scholar] [CrossRef]
- Eisenreich, W.; Joshi, M.; Weber, S.; Bacher, A.; Fischer, M. Natural Abundance Solution 13C NMR Studies of a Phototropin with Photoinduced Polarization. J. Am. Chem. Soc. 2008, 130, 13544–13545. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Joshi, M.; Illarionov, B.; Kacprzak, S.; Lukaschek, M.; Kothe, G.; Budisa, N.; Fischer, M.; Bacher, A.; Weber, S. Strategy for Enhancement of 13C-Photo-CIDNP NMR Spectra by Exploiting Fractional 13C-Labeling of Tryptophan. J. Phys. Chem. B 2015, 119, 13934–13943. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kiryutin, A.S.; Yurkovskaya, A.V.; Sosnovsky, D.V.; Sagdeev, R.Z.; Bannister, S.; Kottke, T.; Kar, R.K.; Schapiro, I.; Ivanov, K.L.; et al. Nuclear Spin-Hyperpolarization Generated in a Flavoprotein under Illumination: Experimental Field-Dependence and Theoretical Level Crossing Analysis. Sci. Rep. 2019, 9, 18436. [Google Scholar] [CrossRef]
- Ding, Y.; Kiryutin, A.S.; Zhao, Z.; Xu, Q.Z.; Zhao, K.H.; Kurle, P.; Bannister, S.; Kottke, T.; Sagdeev, R.Z.; Ivanov, K.L.; et al. Tailored Flavoproteins Acting as Light-Driven Spin Machines Pump Nuclear Hyperpolarization. Sci. Rep. 2020, 10, 18658. [Google Scholar] [CrossRef] [PubMed]
- Thamarath, S.S.; Heberle, J.; Hore, P.J.; Kottke, T.; Matysik, J. Solid-State Photo-CIDNP Effect Observed in Phototropin LOV1-C57S by 13C Magic-Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 15542–15543. [Google Scholar] [CrossRef]
- Pompe, N.; Illarionov, B.; Fischer, M.; Bacher, A.; Weber, S. Completing the Picture: Determination of 13 C Hyperfine Coupling Constants of Flavin Semiquinone Radicals by Photochemically Induced Dynamic Nuclear Polarization Spectroscopy. J. Phys.Chem. Lett. 2022, 5160–5167. [Google Scholar] [CrossRef]
- Zysmilich, M.G.; McDermott, A. Photochemically Induced Dynamic Nuclear Polarization in the Solid-State 15N Spectra of Reaction Centers from Photosynthetic Bacteria Rhodobacter Spbaeroides R-26. J. Am. Chem. Soc. 1994, 116, 8362–8363. [Google Scholar] [CrossRef]
- Daviso, E.; Alia, A.; Prakash, S.; Diller, A.; Gast, P.; Lugtenburg, J.; Matysik, J.; Jeschke, G. Electron-Nuclear Spin Dynamics in a Bacterial Photosynthetic Reaction Center. J. Phys. Chem. C 2009, 113, 10269–10278. [Google Scholar] [CrossRef]
- Jeschke, G. A New Mechanism for Chemically Induced Dynamic Nuclear Polarization in the Solid State. J. Am. Chem. Soc. 1998, 120, 4425–4429. [Google Scholar] [CrossRef]
- Jeschke, G. Electron-Electron-Nuclear Three-Spin Mixing in Spin-Correlated Radical Pairs. J. Chem. Phys. 1997, 106, 10072–10086. [Google Scholar] [CrossRef]
- Polenova, T.; McDermott, A.E. A Coherent Mixing Mechanism Explains the Photoinduced Nuclear Polarization in Photosynthetic Reaction Centers. J. Phys. Chem. B 1999, 103, 535–548. [Google Scholar] [CrossRef]
- McDermott, A.; Zysmilich, M.G.; Polenova, T. Solid State NMR Studies of Photoinduced Polarization in Photosynthetic Reaction Centers: Mechanism and Simulations. Solid State Nucl. Magn. Reson. 1998, 11, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Boxer, S.G.; Chidsey, C.E.D.; Roelofs, M.G. Anisotropic Magnetic Interactions in the Primary Radical Ion-Pair of Photosynthetic Reaction Centers. Proc. Natl. Acad. Sci. USA 1982, 79, 4632–4636. [Google Scholar] [CrossRef]
- Sosnovsky, D.V.; Jeschke, G.; Matysik, J.; Vieth, H.M.; Ivanov, K.L. Level Crossing Analysis of Chemically Induced Dynamic Nuclear Polarization: Towards a Common Description of Liquid-State and Solid-State Cases. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Matysik, J.; Diller, A.; Roy, E.; Alia, A. The Solid-State Photo-CIDNP Effect. Photosynth Res 2009, 102, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Thamarath, S.S.; Bode, B.E.; Prakash, S.; Sai Sankar Gupta, K.B.; Alia, A.; Jeschke, G.; Matysik, J. Electron Spin Density Distribution in the Special Pair Triplet of Rhodobacter Sphaeroides R26 Revealed by Magnetic Field Dependence of the Solid-State Photo-CIDNP Effect. J. Am. Chem. Soc. 2012, 134, 5921–5930. [Google Scholar] [CrossRef]
- Gräsing, D.; Bielytskyi, P.; Céspedes-Camacho, I.F.; Alia, A.; Marquardsen, T.; Engelke, F.; Matysik, J. Field-Cycling NMR with High-Resolution Detection under Magic-Angle Spinning: Determination of Field-Window for Nuclear Hyperpolarization in a Photosynthetic Reaction Center. Sci. Rep. 2017, 7, 12111. [Google Scholar] [CrossRef]
- Thamarath Surendran, S. Towards Photo-CIDNP MAS NMR as a Generally Applicable Enhancement Method. Ph.D. Thesis, Leiden University, Leiden, The Netherlands, 2014. [Google Scholar]
- Bouly, J.P.; Schleicher, E.; Dionisio-Sese, M.; Vandenbussche, F.; Van Der Straeten, D.; Bakrim, N.; Meier, S.; Batschauer, A.; Galland, P.; Bittl, R.; et al. Cryptochrome Blue Light Photoreceptors Are Activated through Interconversion of Flavin Redox States. J. Biol.Chem. 2007, 282, 9383–9391. [Google Scholar] [CrossRef]
- Vaidya, A.T.; Top, D.; Manahan, C.C.; Tokuda, J.M.; Zhang, S.; Pollack, L.; Young, M.W.; Crane, B.R. Flavin Reduction Activates Drosophila Cryptochrome. Proc. Natl. Acad. Sci. USA 2013, 110, 20455–20460. [Google Scholar] [CrossRef]
- Schulten, K.; Swenberg, C.E.; Weiler, A. A Biomagnetic Sensory Mechanism Based on Magnetic Field Modulated Coherent Electron Spin Motion. Z. Phys. Chem. 1978, 111, 1–5. [Google Scholar] [CrossRef]
- Maeda, K.; Henbest, K.B.; Cintolesi, F.; Kuprov, I.; Rodgers, C.T.; Liddell, P.A.; Gust, D.; Timmel, C.R.; Hore, P.J. Chemical Compass Model of Avian Magnetoreception. Nature 2008, 453, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Frederiksen, A.; Hanić, M.; Schuhmann, F.; Grüning, G.; Hore, P.J.; Solov’yov, I.A. Navigation of Migratory Songbirds: A Quantum Magnetic Compass Sensor. Neuroforum 2021, 27, 141–150. [Google Scholar] [CrossRef]
- Solov’Yov, I.A.; Mouritsen, H.; Schulten, K. Acuity of a Cryptochrome and Vision-Based Magnetoreception System in Birds. Biophys. J. 2010, 99, 40–49. [Google Scholar] [CrossRef]
- Solov’yov, I.A.; Schulten, K. Magnetoreception through Cryptochrome May Involve Superoxide. Biophys. J. 2009, 96, 4804–4813. [Google Scholar] [CrossRef] [PubMed]
- Solov’yov, I.A.; Greiner, W. Iron-Mineral-Based Magnetoreceptor in Birds: Polarity or Inclination Compass? Eur. Phys. J. D 2009, 51, 161–172. [Google Scholar] [CrossRef]
- Ritz, T.; Adem, S.; Schulten, K. A Model for Photoreceptor-Based Magnetoreception in Birds. Biophys. J. 2000, 78, 707–718. [Google Scholar] [CrossRef]
- Ritz, T.; Thalau, P.; Phillips, J.B.; Wiltschko, R.; Wiltschko, W. Resonance Effects Indicate a Radical-Pair Mechanism for Avian Magnetic Compass. Nature 2004, 429, 177–180. [Google Scholar] [CrossRef]
- Solov’yov, I.A.; Chandler, D.E.; Schulten, K. Magnetic Field Effects in Arabidopsis Thaliana Cryptochrome-1. Biophys. J. 2007, 92, 2711–2726. [Google Scholar] [CrossRef]
- Pedersen, J.B.; Nielsen, C.; Solov’yov, I.A. Multiscale Description of Avian Migration: From Chemical Compass to Behaviour Modeling. Sci. Rep. 2016, 6, 36709. [Google Scholar] [CrossRef]
- Solov’yov, I.A.; Schulten, K. Reaction Kinetics and Mechanism of Magnetic Field Effects in Cryptochrome. J. Phys. Chem. B 2012, 116, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Grüning, G.; Wong, S.Y.; Gerhards, L.; Schuhmann, F.; Kattnig, D.R.; Hore, P.J.; Solov’yov, I.A. Effects of Dynamical Degrees of Freedom on Magnetic Compass Sensitivity: A Comparison of Plant and Avian Cryptochromes. J. Am. Chem. Soc. 2022, 144, 22902–22914. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Robinson, A.J.; Henbest, K.B.; Hogben, H.J.; Biskup, T.; Ahmad, M.; Schleicher, E.; Weber, S.; Timmel, C.R.; Hore, P.J. Magnetically Sensitive Light-Induced Reactions in Cryptochrome Are Consistent with Its Proposed Role as a Magnetoreceptor. Proc. Natl. Acad. Sci. USA 2012, 109, 4774–4779. [Google Scholar] [CrossRef]
- Kattnig, D.R.; Solov’yov, I.A.; Hore, P.J. Electron Spin Relaxation in Cryptochrome-Based Magnetoreception. Physi.Chem. Chem. Phys. 2016, 18, 12443–12456. [Google Scholar] [CrossRef] [PubMed]
- Hiscock, H.G.; Worster, S.; Kattnig, D.R.; Steers, C.; Jin, Y.; Manolopoulos, D.E.; Mouritsen, H.; Hore, P.J. The Quantum Needle of the Avian Magnetic Compass. Proc. Natl. Acad. Sci. USA 2016, 113, 4634–4639. [Google Scholar] [CrossRef]
- Nielsen, C.; Solov’yov, I.A. MolSpin—Flexible and Extensible General Spin Dynamics Software. J. Chem. Phys. 2019, 151, 194105. [Google Scholar] [CrossRef]
- Lindoy, L.P.; Fay, T.P.; Manolopoulos, D.E. Quantum Mechanical Spin Dynamics of a Molecular Magnetoreceptor. J. Chem. Phys. 2020, 152, 164107. [Google Scholar] [CrossRef]
- Worster, S.; Kattnig, D.R.; Hore, P.J. Spin Relaxation of Radicals in Cryptochrome and Its Role in Avian Magnetoreception. J. Chem. Phys. 2016, 145, 035104. [Google Scholar] [CrossRef]
- Timmel, C.R.; Till, U.; Brocklehurst, B.; Mclauchlan, K.A.; Hore, P.J. Effects of Weak Magnetic Fields on Free Radical Recombination Reactions. Mol. Phys. 1998, 95, 71–89. [Google Scholar] [CrossRef]
- Efimova, O.; Hore, P.J. Role of Exchange and Dipolar Interactions in the Radical Pair Model of the Avian Magnetic Compass. Biophys. J. 2008, 94, 1565–1574. [Google Scholar] [CrossRef]
- Babcock, N.S.; Kattnig, D.R. Radical Scavenging Could Answer the Challenge Posed by Electron–Electron Dipolar Interactions in the Cryptochrome Compass Model. J. Am. Chem. Soc. 2021, 1, 2033–2046. [Google Scholar] [CrossRef]
- Player, T.C.; Hore, P.J. Viability of Superoxide-Containing Radical Pairs as Magnetoreceptors. J. Chem. Phys. 2019, 151, 225101. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Kattnig, D.R.; Sjulstok, E.; Hore, P.J.; Solov’yov, I.A. Ascorbic Acid May Not Be Involved in Cryptochrome-Based Magnetoreception. J. R. Soc. Interface 2017, 14, 20170657. [Google Scholar] [CrossRef]
- Wong, S.Y.; Solov’yov, I.A.; Hore, P.J.; Kattnig, D.R. Nuclear Polarization Effects in Cryptochrome-Based Magnetoreception. J. Chem. Phys. 2021, 154, 035102. [Google Scholar] [CrossRef] [PubMed]
- Bradlaugh, A.A.; Fedele, G.; Munro, A.L.; Hansen, C.N.; Hares, J.M.; Patel, S.; Kyriacou, C.P.; Jones, A.R.; Rosato, E.; Baines, R.A. Essential Elements of Radical Pair Magnetosensitivity in Drosophila. Nature 2023, 615, 111–116. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matysik, J.; Gerhards, L.; Theiss, T.; Timmermann, L.; Kurle-Tucholski, P.; Musabirova, G.; Qin, R.; Ortmann, F.; Solov’yov, I.A.; Gulder, T. Spin Dynamics of Flavoproteins. Int. J. Mol. Sci. 2023, 24, 8218. https://doi.org/10.3390/ijms24098218
Matysik J, Gerhards L, Theiss T, Timmermann L, Kurle-Tucholski P, Musabirova G, Qin R, Ortmann F, Solov’yov IA, Gulder T. Spin Dynamics of Flavoproteins. International Journal of Molecular Sciences. 2023; 24(9):8218. https://doi.org/10.3390/ijms24098218
Chicago/Turabian StyleMatysik, Jörg, Luca Gerhards, Tobias Theiss, Lisa Timmermann, Patrick Kurle-Tucholski, Guzel Musabirova, Ruonan Qin, Frank Ortmann, Ilia A. Solov’yov, and Tanja Gulder. 2023. "Spin Dynamics of Flavoproteins" International Journal of Molecular Sciences 24, no. 9: 8218. https://doi.org/10.3390/ijms24098218
APA StyleMatysik, J., Gerhards, L., Theiss, T., Timmermann, L., Kurle-Tucholski, P., Musabirova, G., Qin, R., Ortmann, F., Solov’yov, I. A., & Gulder, T. (2023). Spin Dynamics of Flavoproteins. International Journal of Molecular Sciences, 24(9), 8218. https://doi.org/10.3390/ijms24098218