

MYCN Amplification Is Associated with Reduced Expression of Genes Encoding γ-Secretase Complex and NOTCH Signaling Components in Neuroblastoma

 ,
,
Abstract
1. Introduction
2. Results
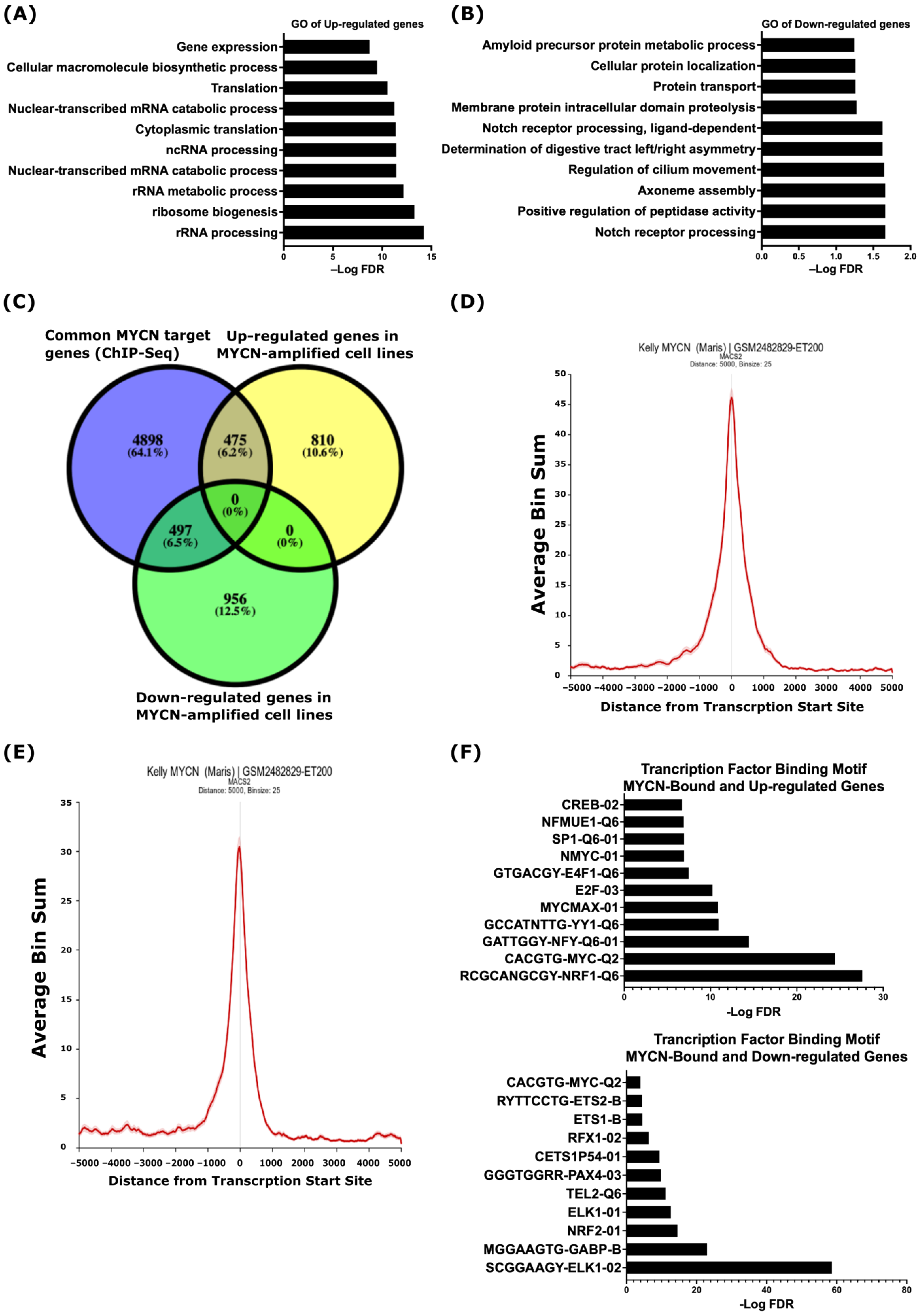
2.1. Identification of MYCN-Regulated Gene Expression Profiles and Biological Processes in MYCN-Amplified Neuroblastoma
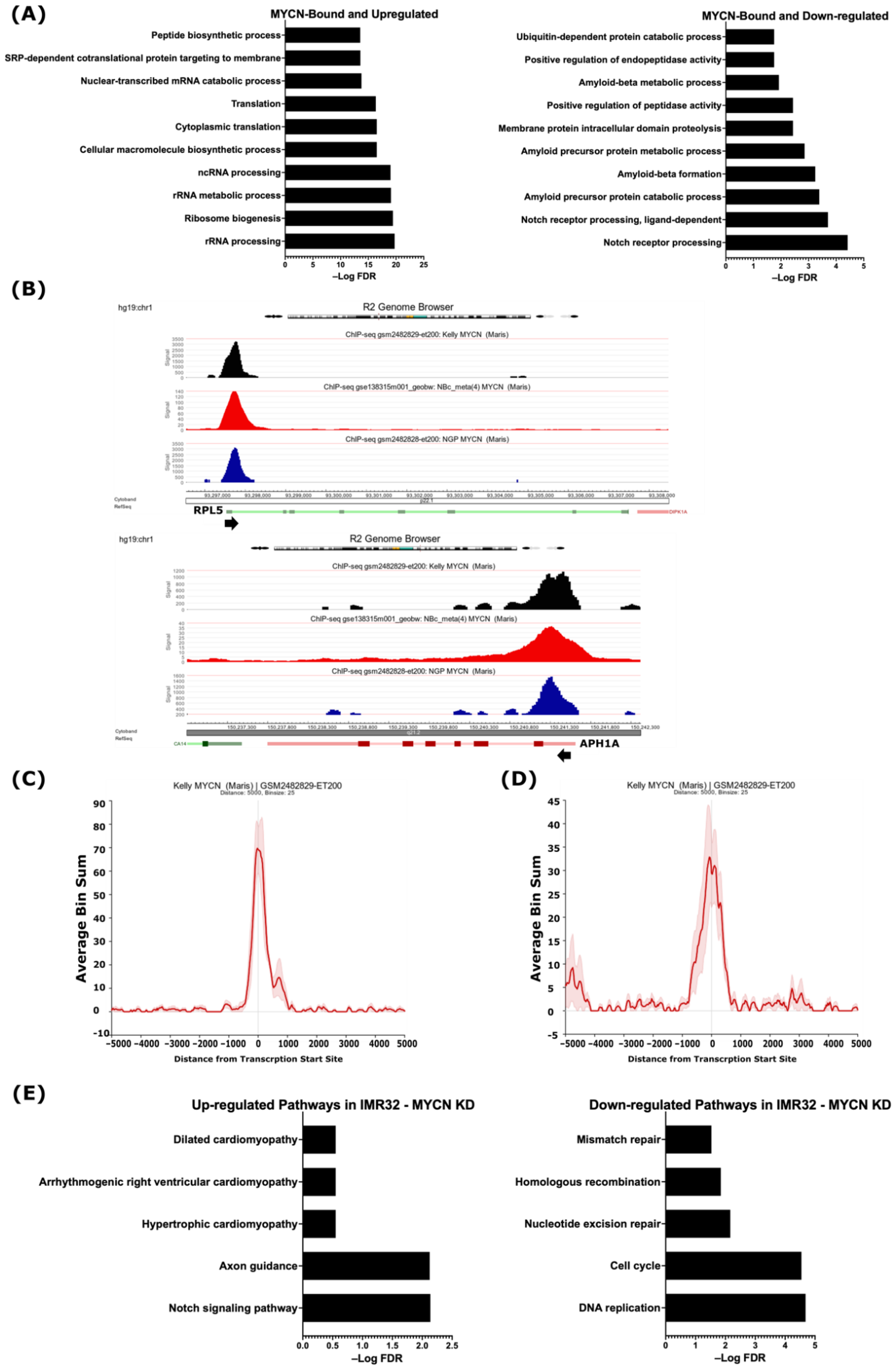
2.2. Integrative Analysis of Gene Expression and MYCN ChIP-Seq Reveals That MYCN Directly Associates with the Suppressed ADAM17 and γ-Secretase Complex Genes
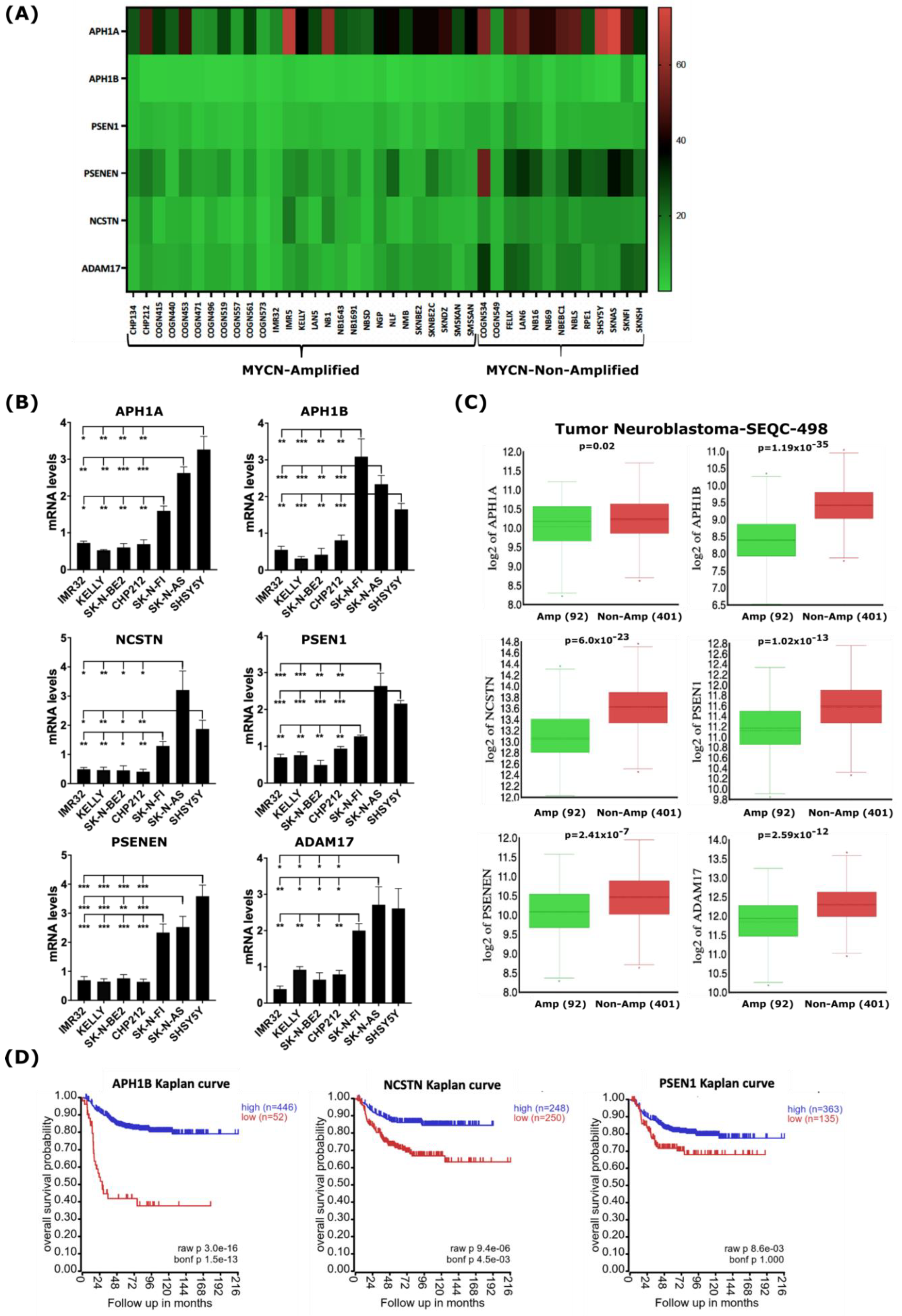
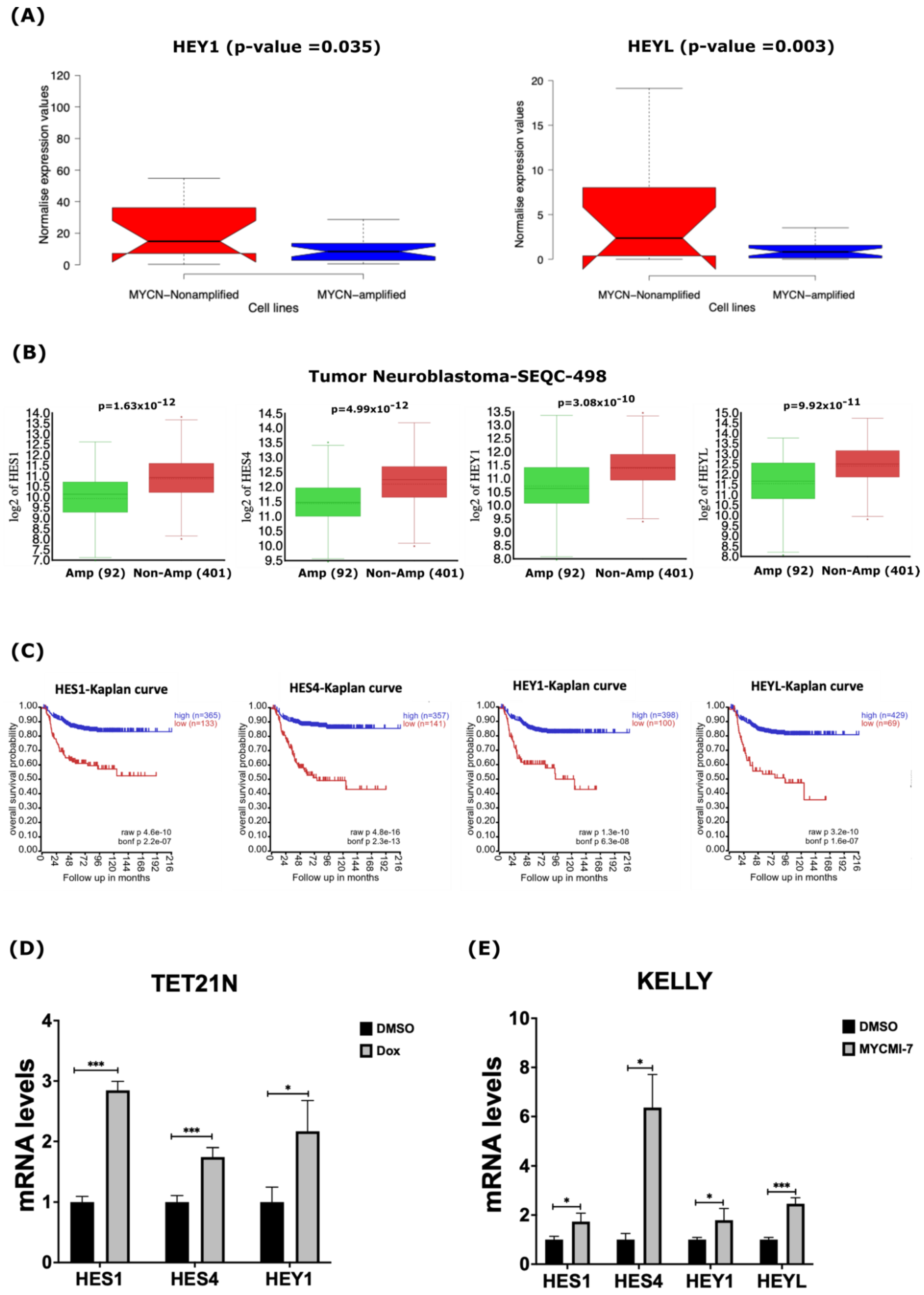
2.3. MYCN Amplification Correlates with Reduced Expression of Genes of the γ-Secretase Complex and ADAM17 in Neuroblastoma
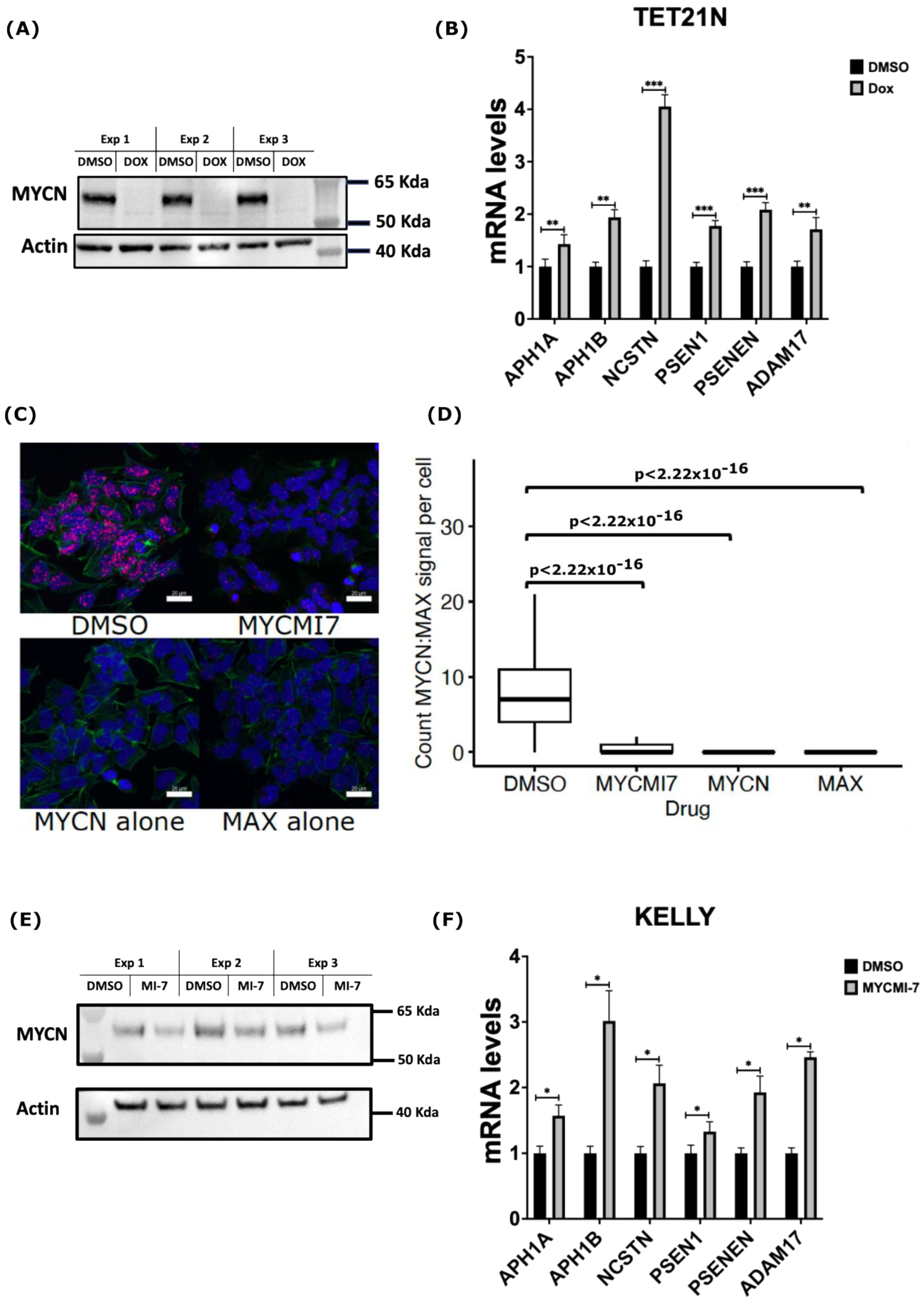
2.4. Genetic and Pharmacological Depletion of MYCN Increases mRNA Levels of γ-Secretase Subunits and ADAM17 in Neuroblastoma
2.5. MYCN Depletion Induces Expression of NOTCH Target Genes in Neuroblastoma
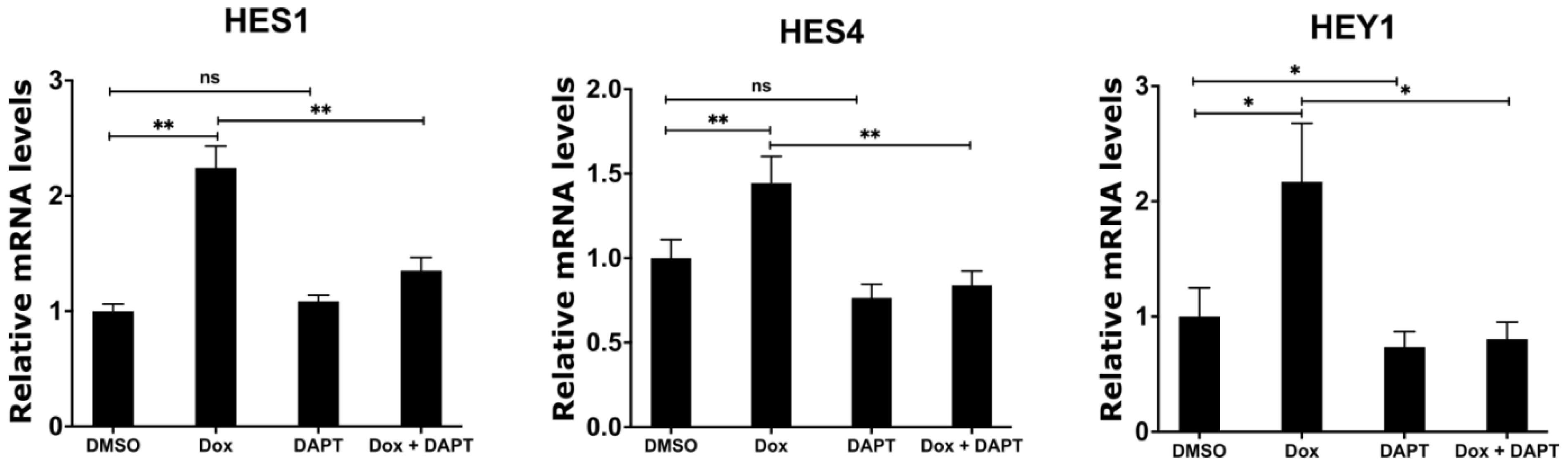
2.6. Inhibition of γ-Secretase Complex Abolishes Induced Expression of NOTCH Target Genes upon MYCN Depletion in Neuroblastoma
3. Discussion
4. Materials and Methods
4.1. RNA-Seq, ChIP-Seq Data, and Gene Ontology (GO) Analysis
4.2. R2: Genomics Analysis and Visualization Platform
4.3. Cell Lines and Cell Cultures
4.4. Resazurin Assay and Assessment of Cell Proliferation and Viability
4.5. RNA Extraction, cDNA Synthesis, and RT-qPCR
4.6. Protein Extraction and Western Blot
4.7. In Situ Proximity Ligation Assay (isPLA)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, N.; De Preter, K.; Hoebeeck, J.; Van Maerken, T.; Pattyn, F.; Mestdagh, P.; Vermeulen, J.; Vandesompele, J.; Speleman, F. The emerging molecular pathogenesis of neuroblastoma: Implications for improved risk assessment and targeted therapy. Genome Med. 2009, 1, 74. [Google Scholar] [CrossRef]
- Newman, E.A.; Nuchtern, J.G. Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin. Pediatr. Surg. 2016, 25, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert. Rev. Anticancer. Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. Embo J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Swier, L.; Dzikiewicz-Krawczyk, A.; Winkle, M.; van den Berg, A.; Kluiver, J. Intricate crosstalk between MYC and non-coding RNAs regulates hallmarks of cancer. Mol. Oncol. 2019, 13, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-specific DNA binding by the c-Myc protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Tesi, A.; de Pretis, S.; Furlan, M.; Filipuzzi, M.; Morelli, M.J.; Andronache, A.; Doni, M.; Verrecchia, A.; Pelizzola, M.; Amati, B.; et al. An early Myc-dependent transcriptional program orchestrates cell growth during B-cell activation. EMBO Rep. 2019, 20, e47987. [Google Scholar] [CrossRef]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massagué, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- Wanzel, M.; Herold, S.; Eilers, M. Transcriptional repression by Myc. Trends Cell Biol. 2003, 13, 146–150. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef]
- McKeown, M.R.; Bradner, J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med. 2014, 4, a014266. [Google Scholar] [CrossRef]
- Castell, A.; Larsson, L.G. Targeting MYC Translation in Colorectal Cancer. Cancer Discov. 2015, 5, 701–703. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [PubMed]
- George, S.L.; Parmar, V.; Lorenzi, F.; Marshall, L.V.; Jamin, Y.; Poon, E.; Angelini, P.; Chesler, L. Novel therapeutic strategies targeting telomere maintenance mechanisms in high-risk neuroblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 78. [Google Scholar] [CrossRef] [PubMed]
- Koneru, B.; Lopez, G.; Farooqi, A.; Conkrite, K.L.; Nguyen, T.H.; Macha, S.J.; Modi, A.; Rokita, J.L.; Urias, E.; Hindle, A.; et al. Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res. 2020, 80, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef]
- Harenza, J.L.; Diamond, M.A.; Adams, R.N.; Song, M.M.; Davidson, H.L.; Hart, L.S.; Dent, M.H.; Fortina, P.; Reynolds, C.P.; Maris, J.M. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci. Data 2017, 4, 170033. [Google Scholar] [CrossRef]
- Upton, K.; Modi, A.; Patel, K.; Kendsersky, N.M.; Conkrite, K.L.; Sussman, R.T.; Way, G.P.; Adams, R.N.; Sacks, G.I.; Fortina, P.; et al. Epigenomic profiling of neuroblastoma cell lines. Sci. Data 2020, 7, 116. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Koster, J.; Haneveld, F.; Aissa, R.A.; van Sluis, P.; Broekmans, M.E.; Molenaar, J.J.; van Nes, J.; Versteeg, R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. USA 2012, 109, 19190–19195. [Google Scholar] [CrossRef] [PubMed]
- Castell, A.; Yan, Q.; Fawkner, K.; Bazzar, W.; Zhang, F.; Wickström, M.; Alzrigat, M.; Franco, M.; Krona, C.; Cameron, D.P.; et al. MYCMI-7: A Small MYC-Binding Compound that Inhibits MYC: MAX Interaction and Tumor Growth in a MYC-Dependent Manner. Cancer Res. Commun. 2022, 2, 182–201. [Google Scholar] [CrossRef]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef]
- Feng, J.; Wang, J.; Liu, Q.; Li, J.; Zhang, Q.; Zhuang, Z.; Yao, X.; Liu, C.; Li, Y.; Cao, L.; et al. DAPT, a γ-Secretase Inhibitor, Suppresses Tumorigenesis, and Progression of Growth Hormone-Producing Adenomas by Targeting Notch Signaling. Front. Oncol. 2019, 9, 809. [Google Scholar] [CrossRef]
- Dovey, H.F.; John, V.; Anderson, J.P.; Chen, L.Z.; de Saint Andrieu, P.; Fang, L.Y.; Freedman, S.B.; Folmer, B.; Goldbach, E.; Holsztynska, E.J.; et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 2001, 76, 173–181. [Google Scholar] [CrossRef]
- Volchenboum, S.L.; Cohn, S.L. Progress in defining and treating high-risk neuroblastoma: Lessons from the bench and bedside. J. Clin. Oncol. 2009, 27, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Saarinen-Pihkala, U.M.; Jahnukainen, K.; Wikström, S.; Koivusalo, A.; Karikoski, R.; Sariola, H.; Hovi, L. Ultrahigh-risk group within the high-risk neuroblastoma category. J. Pediatr. Hematol. Oncol. 2013, 35, e254–e259. [Google Scholar] [CrossRef]
- Pinto, N.; Naranjo, A.; Hibbitts, E.; Kreissman, S.G.; Granger, M.M.; Irwin, M.S.; Bagatell, R.; London, W.B.; Greengard, E.G.; Park, J.R.; et al. Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children’s Oncology Group (COG). Eur. J. Cancer 2019, 112, 66–79. [Google Scholar] [CrossRef]
- Fagnocchi, L.; Zippo, A. Multiple Roles of MYC in Integrating Regulatory Networks of Pluripotent Stem Cells. Front. Cell Dev. Biol. 2017, 5, 7. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Hann, S.R. MYC cofactors: Molecular switches controlling diverse biological outcomes. Cold Spring Harb. Perspect. Med. 2014, 4, a014399. [Google Scholar] [CrossRef]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players, New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.; Dyberg, C.; Pepich, A.; Johnsen, J.I. MYCN Function in Neuroblastoma Development. Front. Oncol. 2020, 10, 624079. [Google Scholar] [CrossRef] [PubMed]
- Gogolin, S.; Dreidax, D.; Becker, G.; Ehemann, V.; Schwab, M.; Westermann, F. MYCN/MYC-mediated drug resistance mechanisms in neuroblastoma. Int. J. Clin. Pharmacol. Ther. 2010, 48, 489–491. [Google Scholar] [CrossRef]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voûte, P.A.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. Embo J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Hald, Ø.H.; Olsen, L.; Gallo-Oller, G.; Elfman, L.H.M.; Løkke, C.; Kogner, P.; Sveinbjörnsson, B.; Flægstad, T.; Johnsen, J.I.; Einvik, C. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 2019, 38, 2800–2813. [Google Scholar] [CrossRef]
- Tao, T.; Sondalle, S.B.; Shi, H.; Zhu, S.; Perez-Atayde, A.R.; Peng, J.; Baserga, S.J.; Look, A.T. The pre-rRNA processing factor DEF is rate limiting for the pathogenesis of MYCN-driven neuroblastoma. Oncogene 2017, 36, 3852–3867. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.; Tran, S.L.; Maritz, M.F.; Liu, B.; Kong, C.F.; Purgato, S.; Yang, C.; Murray, J.; Russell, A.J.; Flemming, C.L.; et al. MYC-Driven Neuroblastomas Are Addicted to a Telomerase-Independent Function of Dyskerin. Cancer Res. 2016, 76, 3604–3617. [Google Scholar] [CrossRef]
- Nakaguro, M.; Kiyonari, S.; Kishida, S.; Cao, D.; Murakami-Tonami, Y.; Ichikawa, H.; Takeuchi, I.; Nakamura, S.; Kadomatsu, K. Nucleolar protein PES1 is a marker of neuroblastoma outcome and is associated with neuroblastoma differentiation. Cancer Sci. 2015, 106, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, C.; Cao, Y.; Liu, L.; Sun, F.; Hou, L. RRS1 knockdown inhibits the proliferation of neuroblastoma cell via PI3K/Akt/NF-κB pathway. Pediatr. Res. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Chang, H.Y.; Chang, J.Y.; Hsu, W.M.; Huang, H.C.; Juan, H.F. Unveiling MYCN regulatory networks in neuroblastoma via integrative analysis of heterogeneous genomics data. Oncotarget 2016, 7, 36293–36310. [Google Scholar] [CrossRef]
- Cortes, C.L.; Veiga, S.R.; Almacellas, E.; Hernández-Losa, J.; Ferreres, J.C.; Kozma, S.C.; Ambrosio, S.; Thomas, G.; Tauler, A. Effect of low doses of actinomycin D on neuroblastoma cell lines. Mol. Cancer 2016, 15, 1. [Google Scholar] [CrossRef]
- Zage, P.E.; Nolo, R.; Fang, W.; Stewart, J.; Garcia-Manero, G.; Zweidler-McKay, P.A. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatr. Blood Cancer 2012, 58, 682–689. [Google Scholar] [CrossRef]
- Van Limpt, V.A.; Chan, A.J.; Van Sluis, P.G.; Caron, H.N.; Van Noesel, C.J.; Versteeg, R. High delta-like 1 expression in a subset of neuroblastoma cell lines corresponds to a differentiated chromaffin cell type. Int. J. Cancer 2003, 105, 61–69. [Google Scholar] [CrossRef]
- Shostak, A.; Ruppert, B.; Ha, N.; Bruns, P.; Toprak, U.H.; Eils, R.; Schlesner, M.; Diernfellner, A.; Brunner, M. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat. Commun. 2016, 7, 11807. [Google Scholar] [CrossRef]
- Iraci, N.; Diolaiti, D.; Papa, A.; Porro, A.; Valli, E.; Gherardi, S.; Herold, S.; Eilers, M.; Bernardoni, R.; Della Valle, G.; et al. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res. 2011, 71, 404–412. [Google Scholar] [CrossRef]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Polycomb group proteins and MYC: The cancer connection. Cell. Mol. Life Sci. 2014, 71, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Corvetta, D.; Chayka, O.; Gherardi, S.; D’Acunto, C.W.; Cantilena, S.; Valli, E.; Piotrowska, I.; Perini, G.; Sala, A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J. Biol. Chem. 2013, 288, 8332–8341. [Google Scholar] [CrossRef]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; Marquez, V.E.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013, 27, 2341–2350. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, P.Y.; Scarlett, C.J.; Malyukova, A.; Liu, B.; Marshall, G.M.; MacKenzie, K.L.; Biankin, A.V.; Liu, T. Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene 2014, 33, 2987–2994. [Google Scholar] [CrossRef]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Viré, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. Embo J. 2005, 24, 336–346. [Google Scholar] [CrossRef]
- Pang, Y.; Liu, J.; Li, X.; Xiao, G.; Wang, H.; Yang, G.; Li, Y.; Tang, S.C.; Qin, S.; Du, N.; et al. MYC and DNMT3A-mediated DNA methylation represses microRNA-200b in triple negative breast cancer. J. Cell Mol. Med. 2018, 22, 6262–6274. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.G.; et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell. 2016, 29, 5–16. [Google Scholar] [CrossRef]
- Sogut, M.S.; Venugopal, C.; Kandemir, B.; Dag, U.; Mahendram, S.; Singh, S.; Gulfidan, G.; Arga, K.Y.; Yilmaz, B.; Kurnaz, I.A. ETS-Domain Transcription Factor Elk-1 Regulates Stemness Genes in Brain Tumors and CD133+ BrainTumor-Initiating Cells. J. Pers. Med. 2021, 11, 125. [Google Scholar] [CrossRef]
- Güner, G.; Lichtenthaler, S.F. The substrate repertoire of γ-secretase/presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hitzenberger, M.; Götz, A.; Menig, S.; Brunschweiger, B.; Zacharias, M.; Scharnagl, C. The dynamics of γ-secretase and its substrates. Semin. Cell Dev. Biol. 2020, 105, 86–101. [Google Scholar] [CrossRef]
- McCaw, T.R.; Inga, E.; Chen, H.; Jaskula-Sztul, R.; Dudeja, V.; Bibb, J.A.; Ren, B.; Rose, J.B. Gamma Secretase Inhibitors in Cancer: A Current Perspective on Clinical Performance. Oncologist 2021, 26, e608–e621. [Google Scholar] [CrossRef] [PubMed]
- López-Nieva, P.; González-Sánchez, L.; Cobos-Fernández, M.; Córdoba, R.; Santos, J.; Fernández-Piqueras, J. More Insights on the Use of γ-Secretase Inhibitors in Cancer Treatment. Oncologist 2021, 26, e298–e305. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Toninelli, G.; Bonini, S.A.; Uberti, D.; Buizza, L.; Bettinsoli, P.; Poliani, P.L.; Facchetti, F.; Memo, M. Targeting Notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro Oncol. 2010, 12, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Grynfeld, A.; Påhlman, S.; Axelson, H. Induced neuroblastoma cell differentiation, associated with transient HES-1 activity and reduced HASH-1 expression, is inhibited by Notch1. Int. J. Cancer 2000, 88, 401–410. [Google Scholar] [CrossRef]
- Chang, H.H.; Lee, H.; Hu, M.K.; Tsao, P.N.; Juan, H.F.; Huang, M.C.; Shih, Y.Y.; Wang, B.J.; Jeng, Y.M.; Chang, C.L.; et al. Notch1 expression predicts an unfavorable prognosis and serves as a therapeutic target of patients with neuroblastoma. Clin. Cancer Res. 2010, 16, 4411–4420. [Google Scholar] [CrossRef]
- Allen, T.D.; Rodriguez, E.M.; Jones, K.D.; Bishop, J.M. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 2011, 71, 6010–6018. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef]
- Ryan, R.J.H.; Petrovic, J.; Rausch, D.M.; Zhou, Y.; Lareau, C.A.; Kluk, M.J.; Christie, A.L.; Lee, W.Y.; Tarjan, D.R.; Guo, B.; et al. A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep. 2017, 21, 784–797. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef]
- Williamson, D.; Schwalbe, E.C.; Hicks, D.; Aldinger, K.A.; Lindsey, J.C.; Crosier, S.; Richardson, S.; Goddard, J.; Hill, R.M.; Castle, J.; et al. Medulloblastoma group 3 and 4 tumors comprise a clinically and biologically significant expression continuum reflecting human cerebellar development. Cell Rep. 2022, 40, 111162. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W.; Stöhr, M.; Schürmann, J.; Wenzel, A.; Löhr, A.; Schwab, M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 1996, 13, 803–812. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Function |
|---|---|---|
| APH1A | Aph-1 Homolog A | Promotes nicastrin and presenilin association in the γ-secretase complex, and mediates γ-secretase interaction with substrates prior to cleavage |
| APH1B | Aph-1 Homolog B | Presenilin-stabilizing cofactor |
| NCSTN | Nicastrin | Cleaves integral membrane proteins, including NOTCH receptors. It is believed to function as a stabilizing cofactor for gamma-secretase complex assembly |
| PSEN1 | Presenilin 1 | Protease, regulates gamma-secretase activity, cleaves the intracellular domain of NOTCH receptors leading to the production of active NOTCH intracellular domain (NICD) |
| PSENEN | Presenilin enhancer | Protease, regulates gamma-secretase activity, cleaves the intracellular domain of NOTCH receptors, leading to the production of active NICD |
| ADAM17 | Disintegrin and metalloprotease domain 17 | Membrane-anchored protease, mediates the initial cleavage of the extracellular domain of NOTCH receptors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, P.; Glowacka, A.; Mahmoud, L.; Bazzar, W.; Larsson, L.-G.; Alzrigat, M. MYCN Amplification Is Associated with Reduced Expression of Genes Encoding γ-Secretase Complex and NOTCH Signaling Components in Neuroblastoma. Int. J. Mol. Sci. 2023, 24, 8141. https://doi.org/10.3390/ijms24098141
Agarwal P, Glowacka A, Mahmoud L, Bazzar W, Larsson L-G, Alzrigat M. MYCN Amplification Is Associated with Reduced Expression of Genes Encoding γ-Secretase Complex and NOTCH Signaling Components in Neuroblastoma. International Journal of Molecular Sciences. 2023; 24(9):8141. https://doi.org/10.3390/ijms24098141
Chicago/Turabian StyleAgarwal, Prasoon, Aleksandra Glowacka, Loay Mahmoud, Wesam Bazzar, Lars-Gunnar Larsson, and Mohammad Alzrigat. 2023. "MYCN Amplification Is Associated with Reduced Expression of Genes Encoding γ-Secretase Complex and NOTCH Signaling Components in Neuroblastoma" International Journal of Molecular Sciences 24, no. 9: 8141. https://doi.org/10.3390/ijms24098141
APA StyleAgarwal, P., Glowacka, A., Mahmoud, L., Bazzar, W., Larsson, L.-G., & Alzrigat, M. (2023). MYCN Amplification Is Associated with Reduced Expression of Genes Encoding γ-Secretase Complex and NOTCH Signaling Components in Neuroblastoma. International Journal of Molecular Sciences, 24(9), 8141. https://doi.org/10.3390/ijms24098141