
Interleukin-1ß Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun
 , , and
, , and 
Abstract
1. Introduction
2. Results
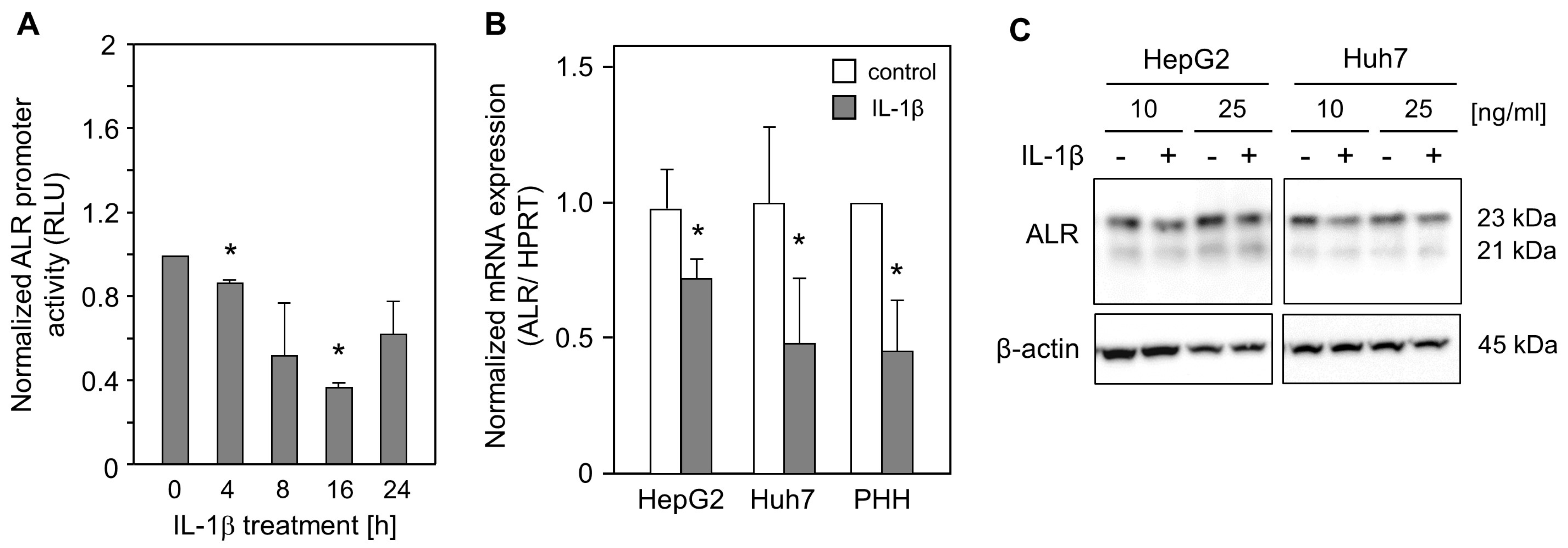
2.1. IL-1ß Reduces ALR Promoter Activity and Expression
2.2. IL-1ß Decreases ALR While Alleviating HNF4α Expression
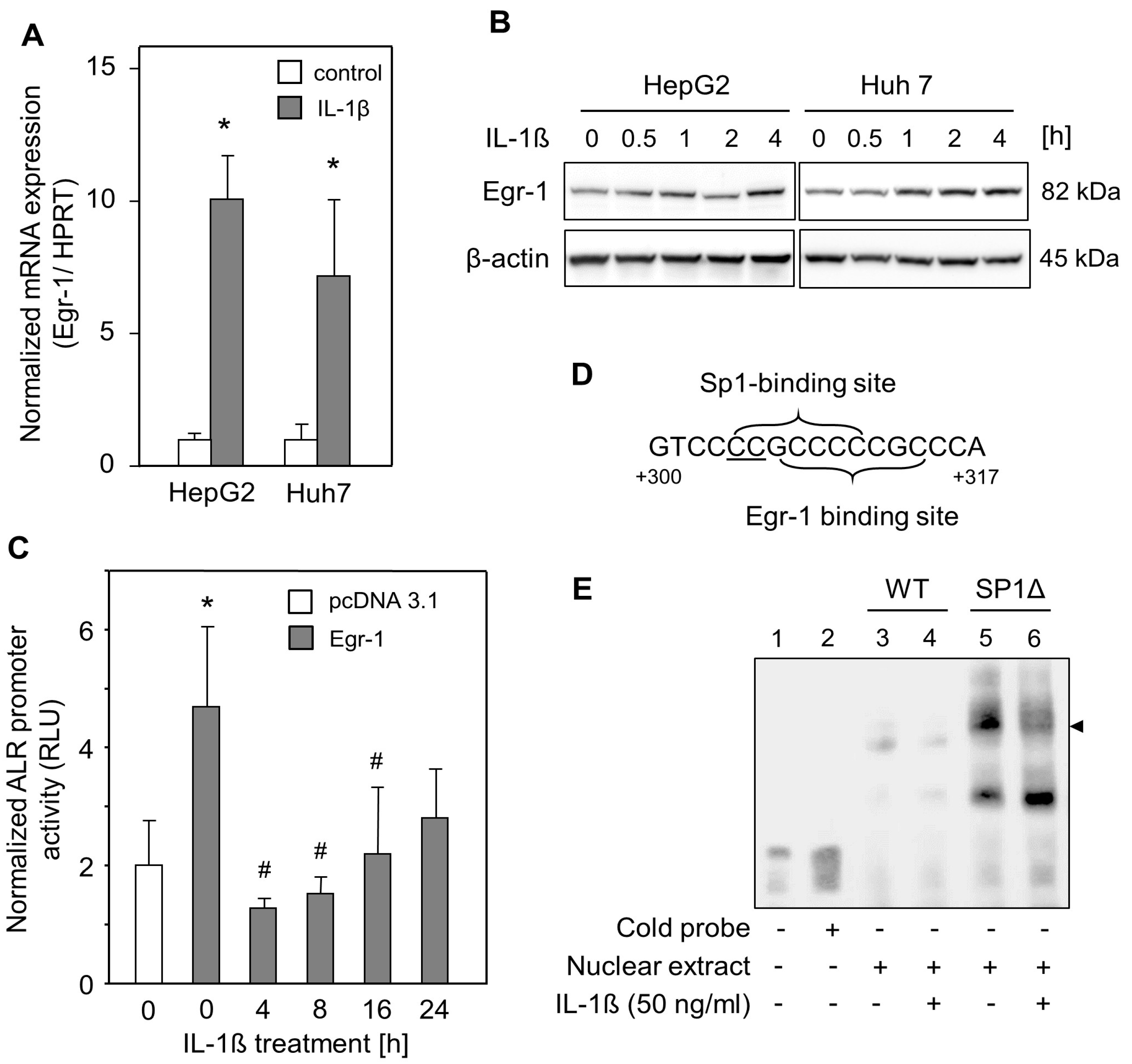
2.3. IL-1ß Reduced Egr-1-Mediated ALR Induction
2.4. Diminished ALR Expression Is Independent of c-Jun Induction by IL-1ß Treatment
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Oxidative stress and the pathogenesis of cholestasis. Semin. Liver Dis. 2010, 30, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jöhrens, K.; Weiss, T.S.; Jonas, W.; Schürmann, A.; Püschel, G.P. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci. Rep. 2018, 8, 16127. [Google Scholar] [CrossRef] [PubMed]
- Donner, M.G.; Schumacher, S.; Warskulat, U.; Heinemann, J.; Häussinger, D. Obstructive cholestasis induces TNF-α- and IL-1β-mediated periportal downregulation of Bsep and zonal regulation of Ntcp, Oatp1a4, and Oatp1b2. Am. J. Physiol. Liver Physiol. 2007, 293, G1134–G1146. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Poupon, R.; Chazouilleres, O.; Poupon, R.E. Chronic cholestatic diseases. J. Hepatol. 2000, 32, 129–140. [Google Scholar] [CrossRef]
- Higuchi, H.; Bronk, S.F.; Taniai, M.; Canbay, A.; Gores, G.J. Cholestasis increases tumor necrosis factor-related apoptotis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes the liver to TRAIL-mediated cytotoxicity. J. Pharmacol. Exp. Ther. 2002, 303, 461–467. [Google Scholar] [CrossRef]
- Sokol, R.J.; Dahl, R.; Devereaux, M.W.; Yerushalmi, B.; Kobak, G.E.; Gumpricht, E. Human Hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J. Craniofacial Surg. 2005, 41, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile Acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef]
- O’Brien, K.M.; Allen, K.M.; Rockwell, C.E.; Towery, K.; Luyendyk, J.P.; Copple, B.L. IL-17A synergistically enhances bile acid–induced inflammation during obstructive cholestasis. Am. J. Pathol. 2013, 183, 1498–1507. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, D.; Weng, J.; Huang, Y.; Liu, S.; Zhang, Q.; Li, N.; Wen, M.; Zhu, G.; Lin, F.; et al. Neutralization of Interleukin-17 Attenuates Cholestatic Liver Fibrosis in Mice. Scand. J. Immunol. 2016, 83, 102–108. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e763. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R. Augmenter of liver regeneration. Fibrogenesis Tissue Repair 2012, 5, 10. [Google Scholar] [CrossRef]
- Thasler, W.E.; Schlott, T.; Thelen, P.; Hellerbrand, C.; Bataille, F.; Lichtenauer, M.; Schlitt, H.-J.; Jauch, K.-W.; Weiss, T.S. Expression of augmenter of liver regeneration (ALR) in human liver cirrhosis and carcinoma. Histopathology 2005, 47, 57–66. [Google Scholar] [CrossRef]
- Cao, Y.; Fu, Y.-L.; Yu, M.; Yue, P.-B.; Ge, C.-H.; Xu, W.-X.; Zhan, Y.-Q.; Li, C.-Y.; Li, W.; Wang, X.-H. Human augmenter of liver regeneration is important for hepatoma cell viability and resistance to radiation-induced oxidative stress. Free. Radic. Biol. Med. 2009, 47, 1057–1066. [Google Scholar] [CrossRef]
- Weiss, T.S.; Lupke, M.; Dayoub, R.; Geissler, E.K.; Schlitt, H.J.; Melter, M.; Eggenhofer, E. Augmenter of Liver Regeneration Reduces Ischemia Reperfusion Injury by Less Chemokine Expression, Gr-1 Infiltration and Oxidative Stress. Cells 2019, 8, 1421. [Google Scholar] [CrossRef]
- Weiss, T.S.; Lupke, M.; Ibrahim, S.; Buechler, C.; Lorenz, J.; Ruemmele, P.; Hofmann, U.; Melter, M.; Dayoub, R. Attenuated lipotoxicity and apoptosis is linked to exogenous and endogenous augmenter of liver regeneration by different pathways. PLoS ONE 2017, 12, e0184282. [Google Scholar] [CrossRef]
- Xiao, W.C.; Zhang, J.; Chen, S.L.; Shi, Y.J.; Xiao, F.; An, W. Alleviation of palmitic acid-induced endoplasmic reticulum stress by augmenter of liver regeneration through IP3R-controlled Ca2+ release. J. Cell. Physiol. 2018, 233, 6148–6157. [Google Scholar] [CrossRef]
- Gandhi, C.R.; Chaillet, J.R.; Nalesnik, M.A.; Kumar, S.; Dangi, A.; Demetris, A.J.; Ferrell, R.; Wu, T.; Divanovic, S.; Stankeiwicz, T.; et al. Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 2015, 148, 379–391.e4. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rani, R.; Karns, R.; Gandhi, C.R. Augmenter of liver regeneration protein deficiency promotes hepatic steatosis by inducing oxidative stress and microRNA-540 expression. FASEB J. 2018, 33, 3825–3840. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Weiss, T.S. Augmenter of liver regeneration: Essential for growth and beyond. Cytokine Growth Factor Rev. 2019, 45, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Sharma, A.; Subramaniyam, N.; Gandhi, C.R. Augmenter of liver regeneration: Mitochondrial function and steatohepatitis. J. Hepatol. 2022, 77, 1410–1421. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, Y.J.; Feng, Y.M.; An, W. The protective roles of augmenter of liver regeneration in hepatocytes in the non-alcoholic fatty liver disease. Front. Pharmacol. 2022, 13, 928606. [Google Scholar] [CrossRef]
- Ibrahim, S.; Dayoub, R.; Krautbauer, S.; Liebisch, G.; Wege, A.K.; Melter, M.; Weiss, T.S. Bile acid-induced apoptosis and bile acid synthesis are reduced by over-expression of Augmenter of Liver Regeneration (ALR) in a STAT3-dependent mechanism. Exp. Cell Res. 2019, 374, 189–197. [Google Scholar] [CrossRef]
- Dayoub, R.; Groitl, P.; Dobner, T.; Bosserhoff, A.K.; Schlitt, H.-J.; Weiss, T.S. Foxa2 (HNF-3β) regulates expression of hepatotrophic factor ALR in liver cells. Biochem. Biophys. Res. Commun. 2010, 395, 465–470. [Google Scholar] [CrossRef]
- Ibrahim, S.; Dayoub, R.; Melter, M.; Weiss, T.S. Bile acids down-regulate the expression of Augmenter of Liver Regeneration (ALR) via SHP/HNF4α1 and independent of Egr-1. Exp. Mol. Pathol. 2018, 105, 236–242. [Google Scholar] [CrossRef]
- Nebbaki, S.S.; El Mansouri, F.E.; Afif, H.; Kapoor, M.; Benderdour, M.; Duval, N.; Pelletier, J.P.; Martel-Pelletier, J.; Fahmi, H. Egr-1 contributes to IL-1-mediated downregulation of peroxisome proliferator-activated receptor gamma expression in human osteoarthritic chondrocytes. Arthritis Res. Ther. 2012, 14, R69. [Google Scholar] [CrossRef]
- Sanchez-Guerrero, E.; Chen, E.; Kockx, M.; An, S.-W.; Chong, B.H.; Khachigian, L.M. Correction: IL-1beta signals through the EGF receptor and activates Egr-1 through MMP-ADAM. PLoS ONE 2013, 7, e39811, Erratum in PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Guo, D.; Dong, L.-Y.; Wu, Y.; Yang, L.; An, W. Down-regulation of hepatic nuclear factor 4α on expression of human hepatic stimulator substance via its action on the proximal promoter in HepG2 cells. Biochem. J. 2008, 415, 111–121. [Google Scholar] [CrossRef]
- Chadjichristos, C.; Ghayor, C.; Kypriotou, M.; Martin, G.; Renard, E.; Ala-Kokko, L.; Suske, G.; de Crombrugghe, B.; Pujol, J.-P.; Galéra, P. Sp1 and Sp3 transcription factors mediate interleukin-1β down-regulation of human type II collagen gene expression in articular chondrocytes. J. Biol. Chem. 2003, 278, 39762–39772. [Google Scholar] [CrossRef]
- Huang, R.P.; Fan, Y.; Ni, Z.; Mercola, D.; Adamson, E.D. Reciprocal modulation between Sp1 and Egr-1. J. Cell. Biochem. 1997, 66, 489–499. [Google Scholar] [CrossRef]
- Dong, L.Y.; Wang, X.N.; Song, Z.G.; Guo, D.; Zhao, Y.Y.; An, W. Identification of human hepatic stimulator substance gene promoter and demonstration of dual regulation of AP1/AP4 cis-acting element in different cell lines. Int. J. Biochem. Cell Biol. 2007, 39, 181–196. [Google Scholar] [CrossRef]
- Simó, R.; Barbosa-Desongles, A.; Hernandez, C.; Selva, D.M. IL1β Down-regulation of Sex Hormone-Binding Globulin Production by Decreasing HNF-4α Via MEK-1/2 and JNK MAPK Pathways. Mol. Endocrinol. 2012, 26, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, R.; Vogel, A.; Schuett, J.; Lupke, M.; Spieker, S.M.; Kettern, N.; Hildt, E.; Melter, M.; Weiss, T.S. Nrf2 activates augmenter of liver regeneration (ALR) via antioxidant response element and links oxidative stress to liver regeneration. Mol. Med. 2013, 19, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Anwar, S.; Almatroudi, A.; Khan, A.A.; Alrumaihi, F.; Alsahli, M.A.; Rahmani, A.H. Hepatoprotective Effects of Garlic Extract against Carbon Tetrachloride (CCl4)-Induced Liver Injury via Modulation of Antioxidant, Anti-Inflammatory Activities and Hepatocyte Architecture. Appl. Sci. 2020, 10, 6200. [Google Scholar] [CrossRef]
- Dayoub, R.; Buerger, L.; Ibrahim, S.; Melter, M.; Weiss, T.S. Augmenter of liver regeneration (ALR) exhibits a dual signaling impact on hepatic acute-phase response. Exp. Mol. Pathol. 2017, 102, 428–433. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Itoh, Y.; Yokomizo, C.; Nishimura, T.; Niimi, T.; Umemura, A.; Fujii, H.; Okanoue, T.; Yoshikawa, T. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-Fed db/db mice. Lab. Investig. 2011, 91, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Svinka, J.; Pflügler, S.; Mair, M.; Marschall, H.-U.; Hengstler, J.G.; Stiedl, P.; Poli, V.; Casanova, E.; Timelthaler, G.; Sibilia, M.; et al. Epidermal growth factor signaling protects from cholestatic liver injury and fibrosis. J. Mol. Med. 2016, 95, 109–117. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Rogers, J.T. Interleukin (IL) 1 beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/I kappa B kinase alpha pathway targeting activator protein-1. J. Biol. Chem. 2008, 283, 25900–25912. [Google Scholar] [CrossRef]
- Hoffmann, E.; Ashouri, J.; Wolter, S.; Doerrie, A.; Dittrich-Breiholz, O.; Schneider, H.; Wagner, E.F.; Troppmair, J.; Mackman, N.; Kracht, M. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J. Biol. Chem. 2008, 283, 12120–12128. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Stepniak, E.; Ricci, R.; Eferl, R.; Sumara, G.; Sumara, I.; Rath, M.; Hui, L.J.; Wagner, E.F. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Gene Dev. 2006, 20, 2306–2314. [Google Scholar] [CrossRef]
- Li, T.G.; Jahan, A.; Chiang, J.Y.L. Bile acids and cytokines inhibit the human cholesterol 7α-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology 2006, 43, 1202–1210. [Google Scholar] [CrossRef]
- Walesky, C.; Gunewardena, S.; Terwilliger, E.F.; Edwards, G.; Borude, P.; Apte, U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4α in adult mice results in increased hepatocyte proliferation. Am. J. Physiol. Liver Physiol. 2013, 304, G26–G37. [Google Scholar] [CrossRef]
- Tan, L.J.; Peng, H.B.; Osaki, M.; Choy, B.K.; Auron, P.E.; Sandell, L.J.; Goldring, M.B. Egr-1 mediates transcriptional repression of COL2A1 promoter activity by interleukin-1 beta. J. Biol. Chem. 2003, 278, 17688–17700. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, A.K.; Rani, R.; Sharma, A.; Wang, J.; Shah, S.A.; Behari, J.; Gonzalez, R.S.; Kohli, R.; Gandhi, C.R. Hepatic deficiency of augmenter of liver regeneration predisposes to nonalcoholic steatohepatitis and fibrosis. Hepatology 2020, 72, 1586–1604. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, H.; Zhao, N.; Zhou, F.; Yang, J. Lipid deposition in liver cells: The influence of short form augmenter of liver regeneration. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 186–194. [Google Scholar] [CrossRef]
- Wei, X.; Murphy, M.A.; Reddy, N.A.; Hao, Y.; Eggertsen, T.G.; Saucerman, J.J.; Bochkis, I.M. Redistribution of lamina-associated domains reshapes binding of pioneer factor FOXA2 in development of nonalcoholic fatty liver disease. Genome Res. 2022, 32, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Sewnath, M.E.; Van Der Poll, T.; Kate, F.J.W.T.; Van Noorden, C.J.F.; Gouma, D.J. Interleukin-1 receptor type I gene-deficient bile duct-ligated mice are partially protected against endotoxin. Hepatology 2002, 35, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Vue, P.M.; Anderson, A.L.; Devereaux, M.W.; Ghosh, S.; Balasubramaniyan, N.; Fillon, S.A.; Dahrenmoeller, C.; Allawzi, A.; Woods, C.; et al. Macrophage-derived IL-1β/NF-κB signaling mediates parenteral nutrition-associated cholestasis. Nat. Commun. 2018, 9, 1393. [Google Scholar] [CrossRef]
- Mutanen, A.; Lohi, J.; Heikkilä, P.; Jalanko, H.; Pakarinen, M.P. Liver Inflammation Relates to Decreased Canalicular Bile Transporter Expression in Pediatric Onset Intestinal Failure. Ann. Surg. 2018, 268, 332–339. [Google Scholar] [CrossRef]
- Cai, S.-Y.; Ge, M.; Mennone, A.; Hoque, R.; Ouyang, X.; Boyer, J.L. Inflammasome Is Activated in the Liver of Cholestatic Patients and Aggravates Hepatic Injury in Bile Duct–Ligated Mouse. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 679–688. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Prelich, J.; Lagoa, C.; Barclay, D.; Zamora, R.; Murase, N.; Gandhi, C.R. Augmenter of liver regeneration (ALR) is a novel biomarker of hepatocellular stress/inflammation: In vitro, in vivo and in silico studies. Mol. Med. 2013, 18, 1421–1429. [Google Scholar] [CrossRef]
- Weiss, T.S.; Dayoub, R. Thy-1 (CD90)-Positive Hepatic Progenitor Cells, Hepatoctyes, and Non-parenchymal Liver Cells Isolated from Human Livers. Methods Mol. Biol. 2017, 1506, 75–89. [Google Scholar] [CrossRef]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef] [PubMed]
- Hadi, N.; Seifati, S.M.; Nateghi, B.; Ravaghi, P.; Khosravian, F.; Namazi, F.; Firouzabad, M.F.; Shaygannejad, V.; Salehi, M. Study of The Correlation between miR-106a, miR-125b, and miR-330 on Multiple Sclerosis Patients by Targeting TNFSF4 and SP1 in NF-eb/TNF-? Pathway: A Case-Control Study. Cell J. 2022, 24, 403–409. [Google Scholar]
- Shi, Q.Q.; Li, J.; Feng, Z.Q.; Zhao, L.C.; Luo, L.; You, Z.M.; Li, D.Y.; Xia, J.; Zuo, G.W.; Chen, D.L. Effect of ginsenoside Rh2 on the migratory ability of HepG2 liver carcinoma cells: Recruiting histone deacetylase and inhibiting activator protein 1 transcription factors. Mol. Med. Rep. 2014, 10, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, Q.-F.; Chang, B.-Y.; Shen, L.-J.; Li, W.; Wu, P.-H.; Fan, W.-J. TFAP4 Promotes Hepatocellular Carcinoma Invasion and Metastasis via Activating the PI3K/AKT Signaling Pathway. Dis. Markers 2019, 2019, 7129214. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALR, sense | 5′-gaagcgggacaccaagttta-3′ | [28] |
| ALR, antisense | 5′-ttcagcacactcctcacagg-3′ | |
| SP1, sense | 5′-ttgaaaaaggagttggtggc-3′ | [62] |
| SP1, antisense | 5′-tgctggttctgtaagttggg-3′ | |
| Egr-1, sense | 5′-tactcctctgttccccctgctt-3′ | |
| Egr-1, antisense | 5′-gaaaaggttgctgtcatgtccg-3′ | |
| HNF4α, sense | 5′-tgtcccgacagatcacctc-3′ | [28] |
| HNF4α, antisense | 5′-cactcaacgagaaccagcag-3′ | |
| c-Jun, sense | 5′-gcaaacctcagcaacttcaacc-3′ | [63] |
| c-Jun, antisense | 5′-gcatctcgggcactgtctga-3′ | |
| AP-4, sense | 5′-gtgcccactcagaaggtgc-3′ | [64] |
| AP-4, antisense | 5′-ggctacagagccctcctatca-3′ | |
| HPRT, sense | 5′-tgacactggcaaaacaatgca-3′ | [28] |
| HPRT, antisense | 5′-ggtccttttcaccagcaagct-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nimphy, J.; Ibrahim, S.; Dayoub, R.; Kubitza, M.; Melter, M.; Weiss, T.S. Interleukin-1ß Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun. Int. J. Mol. Sci. 2023, 24, 8107. https://doi.org/10.3390/ijms24098107
Nimphy J, Ibrahim S, Dayoub R, Kubitza M, Melter M, Weiss TS. Interleukin-1ß Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun. International Journal of Molecular Sciences. 2023; 24(9):8107. https://doi.org/10.3390/ijms24098107
Chicago/Turabian StyleNimphy, Jonas, Sara Ibrahim, Rania Dayoub, Marion Kubitza, Michael Melter, and Thomas S. Weiss. 2023. "Interleukin-1ß Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun" International Journal of Molecular Sciences 24, no. 9: 8107. https://doi.org/10.3390/ijms24098107
APA StyleNimphy, J., Ibrahim, S., Dayoub, R., Kubitza, M., Melter, M., & Weiss, T. S. (2023). Interleukin-1ß Attenuates Expression of Augmenter of Liver Regeneration (ALR) by Regulating HNF4α Independent of c-Jun. International Journal of Molecular Sciences, 24(9), 8107. https://doi.org/10.3390/ijms24098107