The Toxicity of Protein Aggregates: New Insights into the Mechanisms


{kind=link}

Author Contributions
Funding
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Cascella, R.; Chiti, F.; Cecchi, C. Amyloid fibrils act as a reservoir of soluble oligomers, the main culprits in protein deposition diseases. Bioessays 2022, 44, e2200086. [Google Scholar] [CrossRef] [PubMed]
- Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-synuclein aggregated species and their possible roles in disease. Int. J. Mol. Sci. 2020, 21, 8043. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Ermini, E.; Chen, S.W.; Cascella, R.; Cecchi, C. Exploring the release of toxic oligomers from α-synuclein fibrils with Antibodies and STED microscopy. Life 2021, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell. Mol. Life Sci. 2022, 79, 174. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Cascella, R.; Cecchi, C. α-Synuclein oligomers and fibrils: Partners in crime in synucleinopathies. Neural Reg. Res. 2023, 18, 2332–2342. [Google Scholar]
- Boi, L.; Pisanu, A.; Palmas, M.F.; Fusco, G.; Carboni, E.; Casu, M.A.; Satta, V.; Scherma, M.; Janda, E.; Mocci, I.; et al. Modeling Parkinson’s disease neuropathology and symptoms by intranigral inoculation of preformed human α-synuclein oligomers. Int. J. Mol. Sci. 2020, 21, 8535. [Google Scholar] [CrossRef] [PubMed]
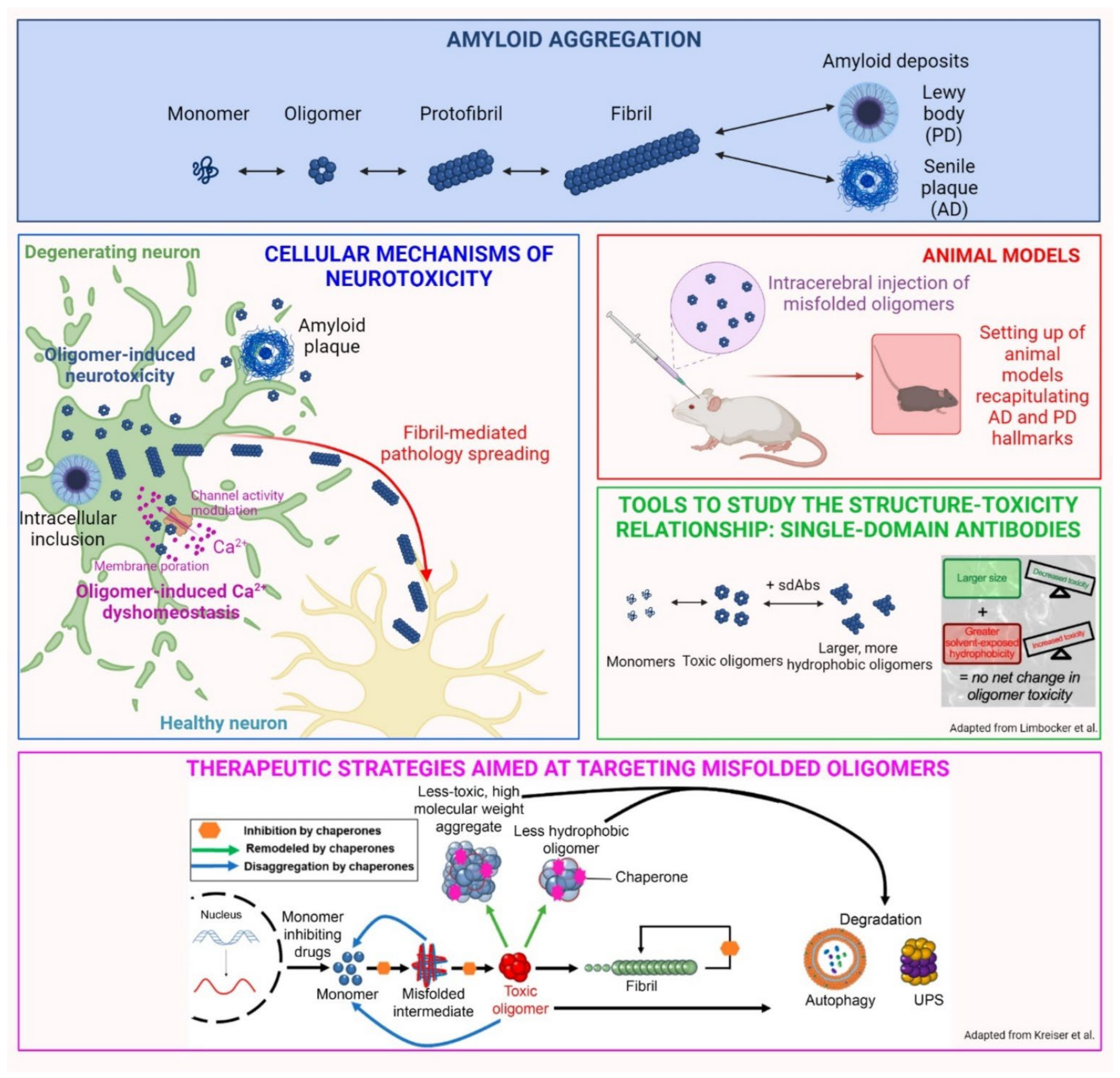
- Limbocker, R.; Mannini, B.; Cataldi, R.; Chhangur, S.; Wright, A.K.; Kreiser, R.P.; Albright, J.A.; Chia, S.; Habchi, J.; Sormanni, P.; et al. Rationally designed antibodies as research tools to study the structure-toxicity relationship of amyloid-β oligomers. Int. J. Mol. Sci. 2020, 21, 4542. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Cecchi, C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Kreiser, R.P.; Wright, A.K.; Block, N.R.; Hollows, J.E.; Nguyen, L.T.; LeForte, K.; Mannini, B.; Vendruscolo, M.; Limbocker, R. Therapeutic strategies to reduce the toxicity of misfolded protein oligomers. Int. J. Mol. Sci. 2020, 21, 8651. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigi, A.; Lombardo, E.; Cascella, R.; Cecchi, C. The Toxicity of Protein Aggregates: New Insights into the Mechanisms. Int. J. Mol. Sci. 2023, 24, 7974. https://doi.org/10.3390/ijms24097974
Bigi A, Lombardo E, Cascella R, Cecchi C. The Toxicity of Protein Aggregates: New Insights into the Mechanisms. International Journal of Molecular Sciences. 2023; 24(9):7974. https://doi.org/10.3390/ijms24097974
Chicago/Turabian StyleBigi, Alessandra, Eva Lombardo, Roberta Cascella, and Cristina Cecchi. 2023. "The Toxicity of Protein Aggregates: New Insights into the Mechanisms" International Journal of Molecular Sciences 24, no. 9: 7974. https://doi.org/10.3390/ijms24097974
APA StyleBigi, A., Lombardo, E., Cascella, R., & Cecchi, C. (2023). The Toxicity of Protein Aggregates: New Insights into the Mechanisms. International Journal of Molecular Sciences, 24(9), 7974. https://doi.org/10.3390/ijms24097974