
SMAD4 Expression in Monocytes as a Potential Biomarker for Atherosclerosis Risk in Patients with Obstructive Sleep Apnea

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
2.1. Subject Characteristics
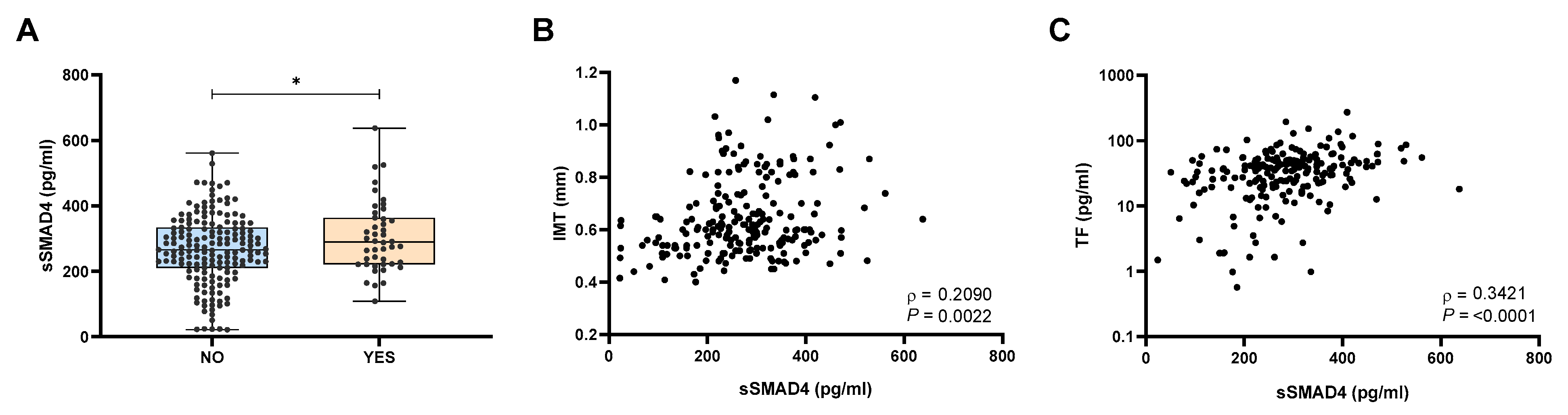
2.2. Patients with Atherosclerosis Exhibited Elevated sSMAD4 Levels
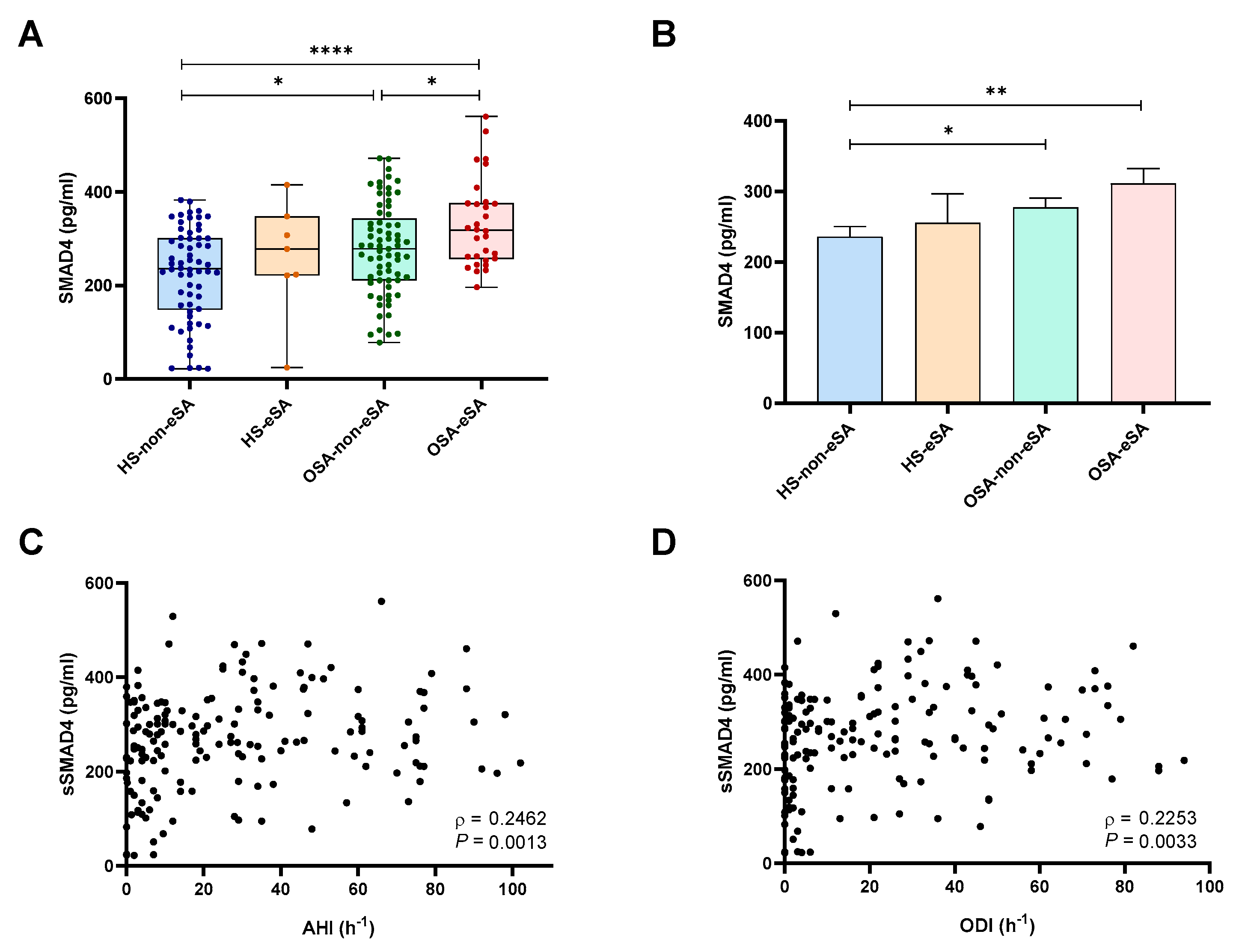
2.3. sSMAD4 Levels Are Elevated in Patients with OSA and eSA and Related to OSA Severity
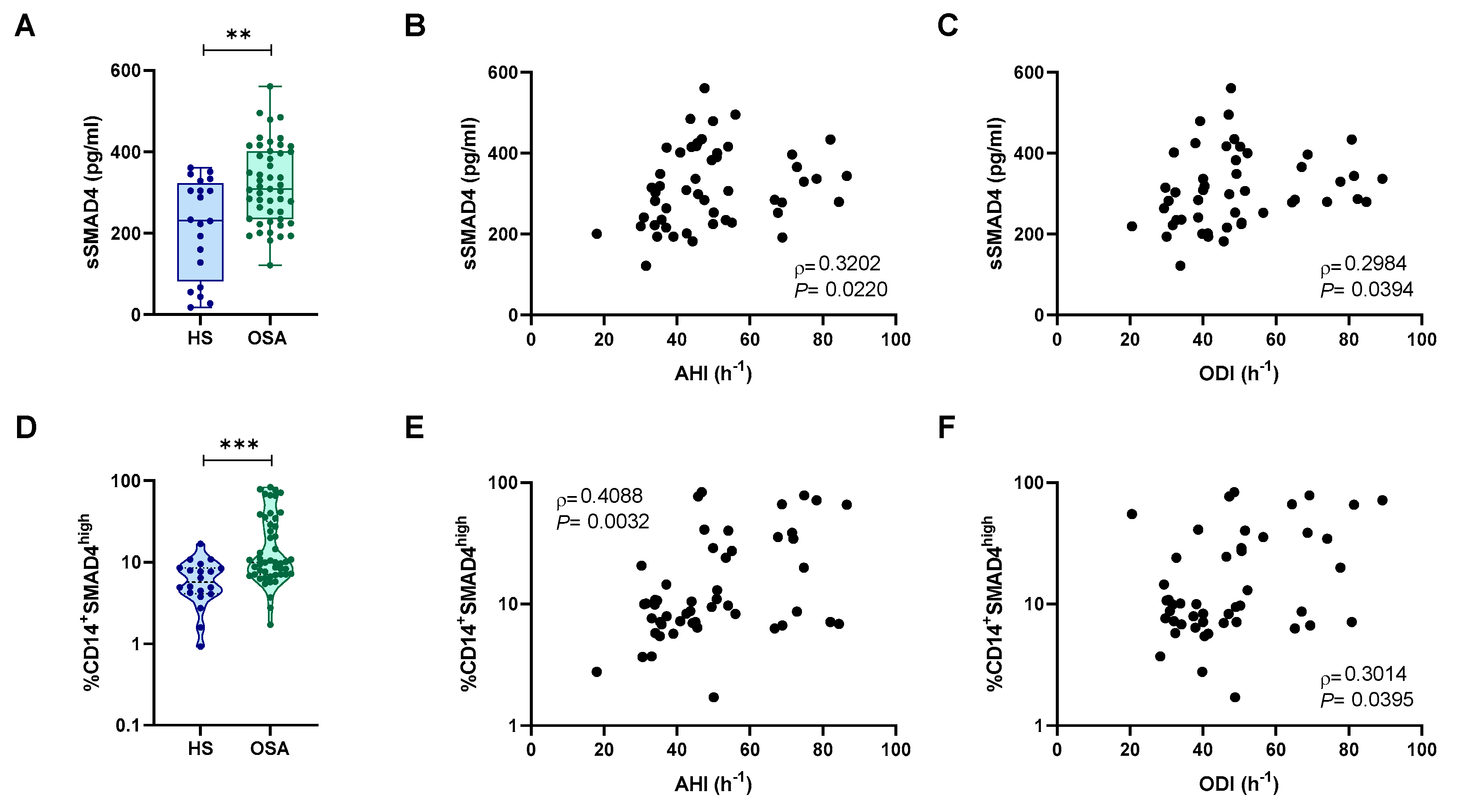
2.4. Intracellular SMAD4 Levels Where Higher in Monocytes from Patients with OSA
2.5. SMAD4 Expression and Release Is Related to Intermittent Hypoxia
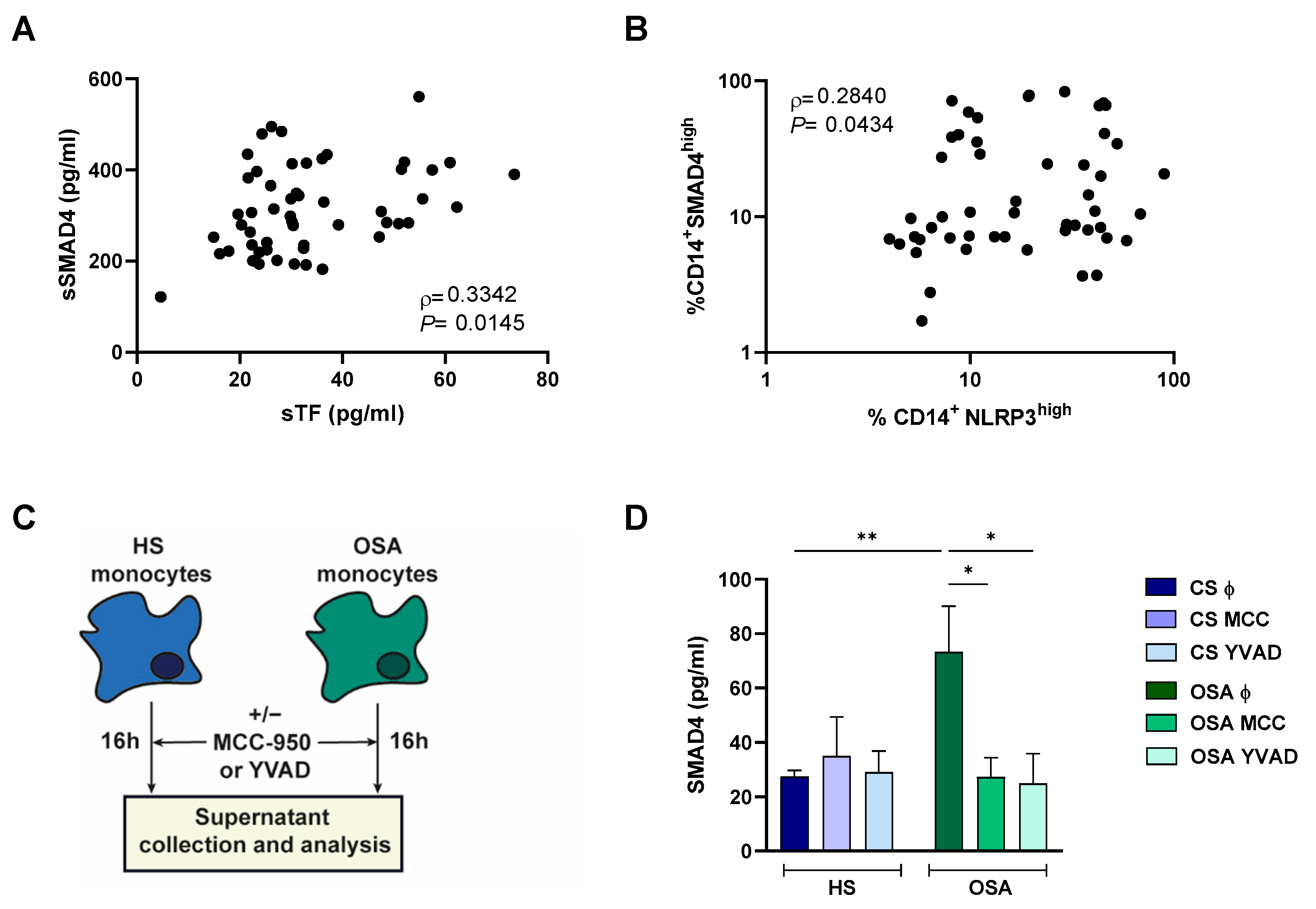
2.6. NLRP3 Inflammasome Could Mediate SMAD4 Release to the Plasma
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Plasma Protein Determination
4.3. Human Cell Isolation
4.4. Flow Cytometry
4.5. mRNA Isolation and Quantification by qPCR
4.6. Intermittent Hypoxia In Vitro Model
4.7. NLRP3 Inhibition Ex Vivo Model
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, P.; Kohler, M.; McNicholas, W.T.; Barbe, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pepin, J.L. Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef]
- Marin, J.M.; Carrizo, S.J.; Vicente, E.; Agusti, A.G. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: An observational study. Lancet 2005, 365, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-de-la-Torre, M.; Campos-Rodriguez, F.; Barbe, F. Obstructive sleep apnoea and cardiovascular disease. Lancet Respir. Med. 2013, 1, 61–72. [Google Scholar] [CrossRef]
- Barros, D.; Garcia-Rio, F. Obstructive sleep apnea and dyslipidemia: From animal models to clinical evidence. Sleep 2019, 42, zsy236. [Google Scholar] [CrossRef]
- Gileles-Hillel, A.; Kheirandish-Gozal, L.; Gozal, D. Biological plausibility linking sleep apnoea and metabolic dysfunction. Nat. Rev. Endocrinol. 2016, 12, 290–298. [Google Scholar] [CrossRef]
- Zinchuk, A.V.; Gentry, M.J.; Concato, J.; Yaggi, H.K. Phenotypes in obstructive sleep apnea: A definition, examples and evolution of approaches. Sleep Med. Rev. 2017, 35, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Heinzer, R.; Vat, S.; Marques-Vidal, P.; Marti-Soler, H.; Andries, D.; Tobback, N.; Mooser, V.; Preisig, M.; Malhotra, A.; Waeber, G.; et al. Prevalence of sleep-disordered breathing in the general population: The HypnoLaus study. Lancet Respir. Med. 2015, 3, 310–318. [Google Scholar] [CrossRef]
- Drager, L.F.; Bortolotto, L.A.; Lorenzi, M.C.; Figueiredo, A.C.; Krieger, E.M.; Lorenzi-Filho, G. Early signs of atherosclerosis in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2005, 172, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.D.; Floras, J.S. Obstructive sleep apnoea and its cardiovascular consequences. Lancet 2009, 373, 82–93. [Google Scholar] [CrossRef]
- Ciccone, M.M.; Scicchitano, P.; Zito, A.; Cortese, F.; Boninfante, B.; Falcone, V.A.; Quaranta, V.N.; Ventura, V.A.; Zucano, A.; Di Serio, F.; et al. Correlation between inflammatory markers of atherosclerosis and carotid intima-media thickness in Obstructive Sleep Apnea. Molecules 2014, 19, 1651–1662. [Google Scholar] [CrossRef]
- Diaz-Garcia, E.; Garcia-Tovar, S.; Alfaro, E.; Jaureguizar, A.; Casitas, R.; Sanchez-Sanchez, B.; Zamarron, E.; Fernandez-Lahera, J.; Lopez-Collazo, E.; Cubillos-Zapata, C.; et al. Inflammasome Activation: A Keystone of Proinflammatory Response in Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2022, 205, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, E.; Sanz-Rubio, D.; Garcia-Tovar, S.; Alfaro, E.; Cubero, P.; Gil, A.V.; Marin, J.M.; Cubillos-Zapata, C.; Garcia-Rio, F. Inflammasome activation mediated by oxidized LDL in patients with sleep apnea and early subclinical atherosclerosis. Eur. Respir. J. 2022, 61, 2201401. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, G.; D’Amico, C.; Vaccaro, V.; D’Anna, M.; Golino, P. The missing link between atherosclerosis, inflammation and thrombosis: Is it tissue factor? Expert Rev. Cardiovasc. Ther. 2011, 9, 517–523. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Martinez-Garcia, M.A.; Diaz-Garcia, E.; Garcia-Tovar, S.; Campos-Rodriguez, F.; Sanchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Pastor, E.; et al. Obstructive sleep apnoea is related to melanoma aggressiveness through paraspeckle protein-1 upregulation. Eur. Respir. J. 2023, 61, 2200707. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef]
- Tabas, I.; Garcia-Cardena, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Toma, I.; McCaffrey, T.A. Transforming growth factor-beta and atherosclerosis: Interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2012, 347, 155–175. [Google Scholar] [CrossRef]
- Bertolino, P.; Deckers, M.; Lebrin, F.; ten Dijke, P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest 2005, 128, 585S–590S. [Google Scholar] [CrossRef]
- van Royen, N.; Hoefer, I.; Buschmann, I.; Heil, M.; Kostin, S.; Deindl, E.; Vogel, S.; Korff, T.; Augustin, H.; Bode, C.; et al. Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J. 2002, 16, 432–434. [Google Scholar] [CrossRef]
- Grainger, D.J.; Byrne, C.D.; Witchell, C.M.; Metcalfe, J.C. Transforming growth factor beta is sequestered into an inactive pool by lipoproteins. J. Lipid Res. 1997, 38, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, E.; Garcia-Tovar, S.; Casitas, R.; Jaureguizar, A.; Zamarron, E.; Sanchez-Sanchez, B.; Sastre-Perona, A.; Lopez-Collazo, E.; Garcia-Rio, F.; Cubillos-Zapata, C. Intermittent Hypoxia Mediates Paraspeckle Protein-1 Upregulation in Sleep Apnea. Cancers 2021, 13, 3888. [Google Scholar] [CrossRef]
- Hernandez-Jimenez, E.; Cubillos-Zapata, C.; Toledano, V.; Perez de Diego, R.; Fernandez-Navarro, I.; Casitas, R.; Carpio, C.; Casas-Martin, J.; Valentin, J.; Varela-Serrano, A.; et al. Monocytes inhibit NK activity via TGF-beta in patients with obstructive sleep apnoea. Eur. Respir. J. 2017, 49, 1602456. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Moren, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.; Hasegawa, H.; Scott, A.; Zhou, X.; Wagner, A.K.; Han, B.X.; Wang, F. Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proc. Natl. Acad. Sci. Am. 2011, 108, 3395–3400. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.E.; Michot, C.; Cormier-Daire, V.; L’Ecuyer, T.J.; Matherne, G.P.; Barnes, B.H.; Humberson, J.B.; Edmondson, A.C.; Zackai, E.; O’Connor, M.J.; et al. Gain-of-function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. Am. J. Med. Genet. A 2016, 170, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.-C.; Liu, X.-M.; Liang, L.-R.; Wang, L.-F.; Zhong, J.-C. Targeting the microRNA-34a as a Novel Therapeutic Strategy for Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 8, 784044. [Google Scholar] [CrossRef]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, E.; Jaureguizar, A.; Casitas, R.; Garcia-Tovar, S.; Sanchez-Sanchez, B.; Zamarron, E.; Lopez-Collazo, E.; Garcia-Rio, F.; Cubillos-Zapata, C. SMAD4 Overexpression in Patients with Sleep Apnoea May Be Associated with Cardiometabolic Comorbidities. J. Clin. Med. 2020, 9, 2378. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front. Cardiovasc. Med. 2022, 9, 959285. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar] [CrossRef]
- Zhao, T.; Ren, H.; Jia, L.; Chen, J.; Xin, W.; Yan, F.; Li, J.; Wang, X.; Gao, S.; Qian, D.; et al. Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ceron, E.; Casitas, R.; Galera, R.; Sanchez-Sanchez, B.; Zamarron, E.; Garcia-Sanchez, A.; Jaureguizar, A.; Cubillos-Zapata, C.; Garcia-Rio, F. Contribution of sleep characteristics to the association between obstructive sleep apnea and dyslipidemia. Sleep Med. 2021, 84, 63–72. [Google Scholar] [CrossRef]
- Nadeem, R.; Singh, M.; Nida, M.; Waheed, I.; Khan, A.; Ahmed, S.; Naseem, J.; Champeau, D. Effect of obstructive sleep apnea hypopnea syndrome on lipid profile: A meta-regression analysis. J. Clin. Sleep Med. 2014, 10, 475–489. [Google Scholar] [CrossRef]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of Lipid Accumulation and Inflammation in Atherosclerosis: Focus on Molecular and Cellular Mechanisms. Front. Cardiovasc. Med. 2021, 8, 707529. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. The epigenetic memory of monocytes and macrophages as a novel drug target in atherosclerosis. Clin. Ther. 2015, 37, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Pardali, E.; Ten Dijke, P. TGFbeta signaling and cardiovascular diseases. Int. J. Biol. Sci. 2012, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Schunkert, H.; Konig, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.; Barbalic, M.; Gieger, C.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef]
- McCaffrey, T.A. TGF-beta signaling in atherosclerosis and restenosis. Front. Biosci. 2009, 1, 236–245. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Liu, Y.; Lao, M.; Chen, J.; Lu, M.; Luo, S.; Ou, Q.; Luo, Z.; Yuan, P.; Chen, J.; Ye, G.; et al. Short-term prognostic effects of circulating regulatory T-Cell suppressive function and vascular endothelial growth factor level in patients with non-small cell lung cancer and obstructive sleep apnea. Sleep Med. 2020, 70, 88–96. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Martinez-Garcia, M.A.; Diaz-Garcia, E.; Jaureguizar, A.; Campos-Rodriguez, F.; Sanchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Pastor, E.; et al. Obesity attenuates the effect of sleep apnea on active TGF-ss1 levels and tumor aggressiveness in patients with melanoma. Sci. Rep. 2020, 10, 15528. [Google Scholar] [CrossRef]
- In, S.M.; Park, D.Y.; Lee, K.I.; Gu, G.; Kim, H.J. The effects of intermittent hypoxia on human nasal mucosa. Sleep Breath. 2021, 25, 1453–1460. [Google Scholar] [CrossRef]
- Loboda, A.; Sobczak, M.; Jozkowicz, A.; Dulak, J. TGF-beta1/Smads and miR-21 in Renal Fibrosis and Inflammation. Mediat. Inflamm. 2016, 2016, 8319283. [Google Scholar] [CrossRef]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Chen, C.; Lei, W.; Chen, W.; Zhong, J.; Gao, X.; Li, B.; Wang, H.; Huang, C. Serum TGF-beta1 and SMAD3 levels are closely associated with coronary artery disease. BMC Cardiovasc. Disord. 2014, 14, 18. [Google Scholar] [CrossRef]
- Guo, X.; Li, M.; Wang, X.; Pan, Y.; Li, J. Correlation between loss of Smad4 and clinical parameters of non-small cell lung cancer: An observational cohort study. BMC Pulm. Med. 2021, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Shan, W.; Ding, X.; Yang, P.; Rozjan, A.; Yao, Q. Intermittent hypoxia induces tumor immune escape in murine S180 solid tumors via the upregulation of TGF-beta(1) in mice. Sleep Breath. 2021, 25, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights into the NLRP 3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Li, N.; Geng, C.; Hou, S.; Fan, H.; Gong, Y. Damage-Associated Molecular Patterns and Their Signaling Pathways in Primary Blast Lung Injury: New Research Progress and Future Directions. Int. J. Mol. Sci. 2020, 21, 6303. [Google Scholar] [CrossRef]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- Faure, E.; Thomas, L.; Xu, H.; Medvedev, A.; Equils, O.; Arditi, M. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: Role of NF-kappa B activation. J. Immunol. 2001, 166, 2018–2024. [Google Scholar] [CrossRef]
- Falck-Hansen, M.; Kassiteridi, C.; Monaco, C. Toll-like receptors in atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008–14023. [Google Scholar] [CrossRef]
- Shinohara, M.; Hirata, K.; Yamashita, T.; Takaya, T.; Sasaki, N.; Shiraki, R.; Ueyama, T.; Emoto, N.; Inoue, N.; Yokoyama, M.; et al. Local overexpression of toll-like receptors at the vessel wall induces atherosclerotic lesion formation: Synergism of TLR2 and TLR4. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.M.; Artal, J.; Martin, T.; Carrizo, S.J.; Andrés, M.; Burriel, I.M.; Bolea, R.; Sanz, A.; Varona, L.; Godino, J.; et al. Epigenetics modifications and Subclinical Atherosclerosis in Obstructive Sleep Apnea: The EPIOSA study. BMC Pulm. Med. 2014, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.B.; Budhiraja, R.; Gottlieb, D.J.; Gozal, D.; Iber, C.; Kapur, V.K.; Marcus, C.L.; Mehra, R.; Parthasarathy, S.; Quan, S.F.; et al. Rules for Scoring Respiratory Events in Sleep: Update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J. Clin. Sleep Med. 2012, 8, 597–619. [Google Scholar] [CrossRef] [PubMed]
- Muntendam, P.; McCall, C.; Sanz, J.; Falk, E.; Fuster, V. The BioImage Study: Novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease—Study design and objectives. Am. Heart J. 2010, 160, 49–57.e1. [Google Scholar] [CrossRef] [PubMed]
- Jarauta, E.; Mateo-Gallego, R.; Bea, A.; Burillo, E.; Calmarza, P.; Civeira, F. Carotid Intima-Media Thickness in Subjects with No Cardiovascular Risk Factors. Rev. Esp. Cardiol. (Engl. Ed.) 2010, 63, 97–102. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects with Atherosclerotic Plaques | Subjects without Atherosclerotic Plaques | |||||||
|---|---|---|---|---|---|---|---|---|
| Overall | Overall | p-Value | Healthy Subjects | OSA Patients | p-Value | |||
| Non-Early Subclinical Atherosclerosis (HS-non-eSA) | Early Subclinical Atherosclerosis (HS-eSA) | Non-Early Subclinical Atherosclerosis (OSA-non-eSA) | Early Subclinical Atherosclerosis (OSA-eSA) | |||||
| n | 44 | 168 | - | 62 | 7 | 69 | 30 | - |
| Male sex, n (%) | 36 (81) | 114 (67) | 0.070 | 35 (56) | 5 (71) | 52 (75) | 22 (73) | 0.08 |
| Age, years | 51 (45–57) | 43 (36–52) | 0.001 | 36 (33–43) | 45 (41–51) * | 43 (38–58) # | 53 (47–59) * | <0.0001 |
| Body mass index, kg/m2 | 30.2 (27.3–33.3) | 29.5 (26.5–33.9) | 0.890 | 27.4 (24.5–30) | 30.2 (27.1–32.8) | 30.1 (27.2–34.6) # | 32.3 (29.8–37.1) | <0.0001 |
| Body fat, % | 32.3 (28.6–37.1) | 32.8 (27.2–40) | 0.458 | 32 (23.3–40.7) | 34.6 (27.7–42.1) | 33.9 (27.8–39.5) # | 32.6 (28.6–39.6) # | 0.537 |
| Current smoker, n (%) | 11 (25) | 37 (22) | 0.675 | 12 (19) | 0 (0) | 16 (23) | 9 (30) | 0.334 |
| Former smoker, n (%) | 18 (41) | 45 (27) | 0.070 | 18 (29) | 3 (43) | 16 (23) | 8 (27) | 0.674 |
| Pack-years | 15 (0–30) | 0 (0–14) | 0.002 | 0 (0–14) | 0 (0–19) | 0 (0–11) | 7 (0–27) | 0.174 |
| Epworth Sleepiness Scale | 8 (5–12) | 10 (6–13) | 0.108 | 9 (6–13) | 10 (8–14) | 14 (2.5–33) | 20 (6.7–46.2) | 0.329 |
| AHI, events/h | 32.5 (14.8–61.5) | 18 (5–45.6) | 0.012 | 4 (1.2–7.2) | 4 (2–8) | 35.0 (21.5–62.5) # | 42 (27.7–59.2) # | <0.0001 |
| ODI, events/h | 35.5 (15.7–51.2) | 21 (2–43) | 0.036 | 0 (0–2) | 2.5 (1.5–3.5) | 29 (17–49) # | 40 (26–60) # | <0.0001 |
| CT90, % | 14 (2–35.3) | 5 (0–20.5) | 0.018 | 0 (0–2) | 0 (0–1) | 10 (1.5–25) # | 20 (6–43.5) # | <0.0001 |
| Mean nocturnal SaO2, % | 92.5 (91–94) | 94 (92–95) | 0.100 | 95 (94–96) | 94 (94–95) | 93 (91–94) # | 92 (90–93) | <0.0001 |
| Low nocturnal SaO2, % | 78 (71–85) | 83 (75–88) | 0.018 | 88 (84–92) | 89 (83–92) | 79 (71–84) # | 77 (70–82) # | <0.0001 |
| Systolic BP, mm Hg | 133 (128–144) | 126 (116–134) | 0.0002 | 120 (111–131) | 124 (115–130) | 127 (120–134) | 132 (123–145) | 0.0002 |
| Diastolic BP, mm Hg | 85 (76–94) | 80 (71–87) | 0.029 | 74 (68–82) | 81 (73–91) | 85 (76–91) # | 83 (75–95) | 0.0001 |
| IMT, mm | 0.65 (0.58–0.76) | 0.59 (0.53–0.72) | 0.042 | 0.53 (0.50–0.57) | 0.86 (0.82–0.90) * | 0.59 (0.54–0.63) # | 0.86 (0.82–0.90) * | <0.0001 |
| Non-Apneic Healthy Subjects (n = 20) | Severe OSA Patients (n = 50) | p-Value | |
|---|---|---|---|
| Male sex, n (%) | 12 (60) | 44 (73) | 0.260 |
| Age, years | 51 (47–58) | 58 (51–64) | 0.176 |
| Body mass index, kg/m2 | 28.3 (25.7–34.5) | 31.2 (28.4–33.8) | 0.105 |
| Current smoker n (%) | 4 (20) | 9 (15) | 0.433 |
| Former smoker n (%) | 2 (10) | 11 (18.3) | 0.363 |
| Epworth Sleepiness Scale | 2 (0–4) | 8 (5–13) | <0.001 |
| AHI, events/h | 3.2 (1.1–5.8) | 47.5 (36.4–67.25) | <0.001 |
| ODI, events/h | 3 (0–5.2) | 46 (33.9–62.5) | <0.001 |
| Mean nocturnal SaO2, % | 94 (93–96) | 90 (88–92) | <0.001 |
| Low nocturnal SaO2, % | 89 (82–93) | 75 (65.5–80) | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-García, E.; García-Sánchez, A.; Sánz-Rubio, D.; Alfaro, E.; López-Fernández, C.; Casitas, R.; Mañas Baena, E.; Cano-Pumarega, I.; Cubero, P.; Marin-Oto, M.; et al. SMAD4 Expression in Monocytes as a Potential Biomarker for Atherosclerosis Risk in Patients with Obstructive Sleep Apnea. Int. J. Mol. Sci. 2023, 24, 7900. https://doi.org/10.3390/ijms24097900
Díaz-García E, García-Sánchez A, Sánz-Rubio D, Alfaro E, López-Fernández C, Casitas R, Mañas Baena E, Cano-Pumarega I, Cubero P, Marin-Oto M, et al. SMAD4 Expression in Monocytes as a Potential Biomarker for Atherosclerosis Risk in Patients with Obstructive Sleep Apnea. International Journal of Molecular Sciences. 2023; 24(9):7900. https://doi.org/10.3390/ijms24097900
Chicago/Turabian StyleDíaz-García, Elena, Aldara García-Sánchez, David Sánz-Rubio, Enrique Alfaro, Cristina López-Fernández, Raquel Casitas, Eva Mañas Baena, Irene Cano-Pumarega, Pablo Cubero, Marta Marin-Oto, and et al. 2023. "SMAD4 Expression in Monocytes as a Potential Biomarker for Atherosclerosis Risk in Patients with Obstructive Sleep Apnea" International Journal of Molecular Sciences 24, no. 9: 7900. https://doi.org/10.3390/ijms24097900
APA StyleDíaz-García, E., García-Sánchez, A., Sánz-Rubio, D., Alfaro, E., López-Fernández, C., Casitas, R., Mañas Baena, E., Cano-Pumarega, I., Cubero, P., Marin-Oto, M., López-Collazo, E., Marin, J. M., García-Río, F., & Cubillos-Zapata, C. (2023). SMAD4 Expression in Monocytes as a Potential Biomarker for Atherosclerosis Risk in Patients with Obstructive Sleep Apnea. International Journal of Molecular Sciences, 24(9), 7900. https://doi.org/10.3390/ijms24097900