Identification of Protein Complexes by Integrating Protein Abundance and Interaction Features Using a Deep Learning Strategy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
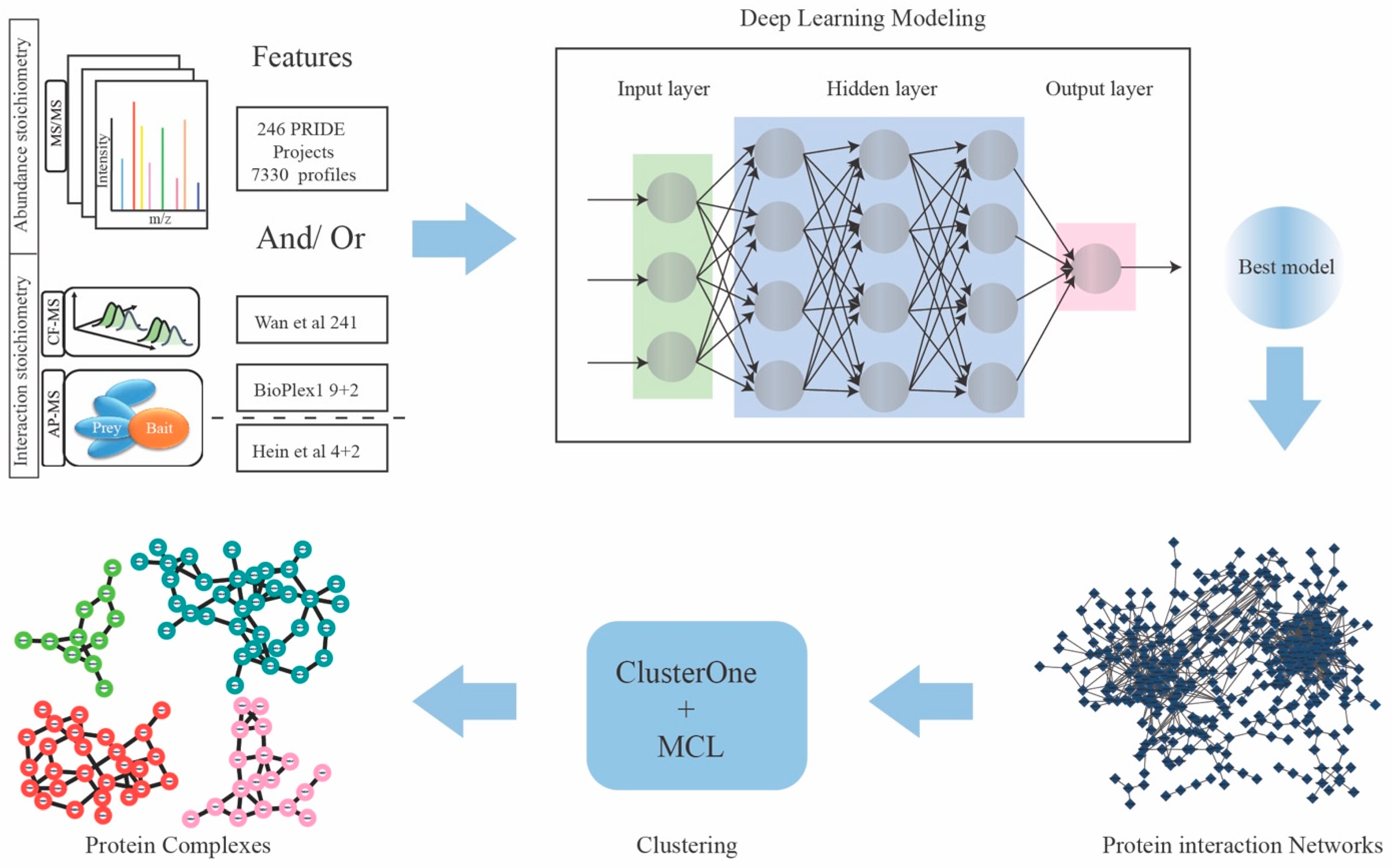
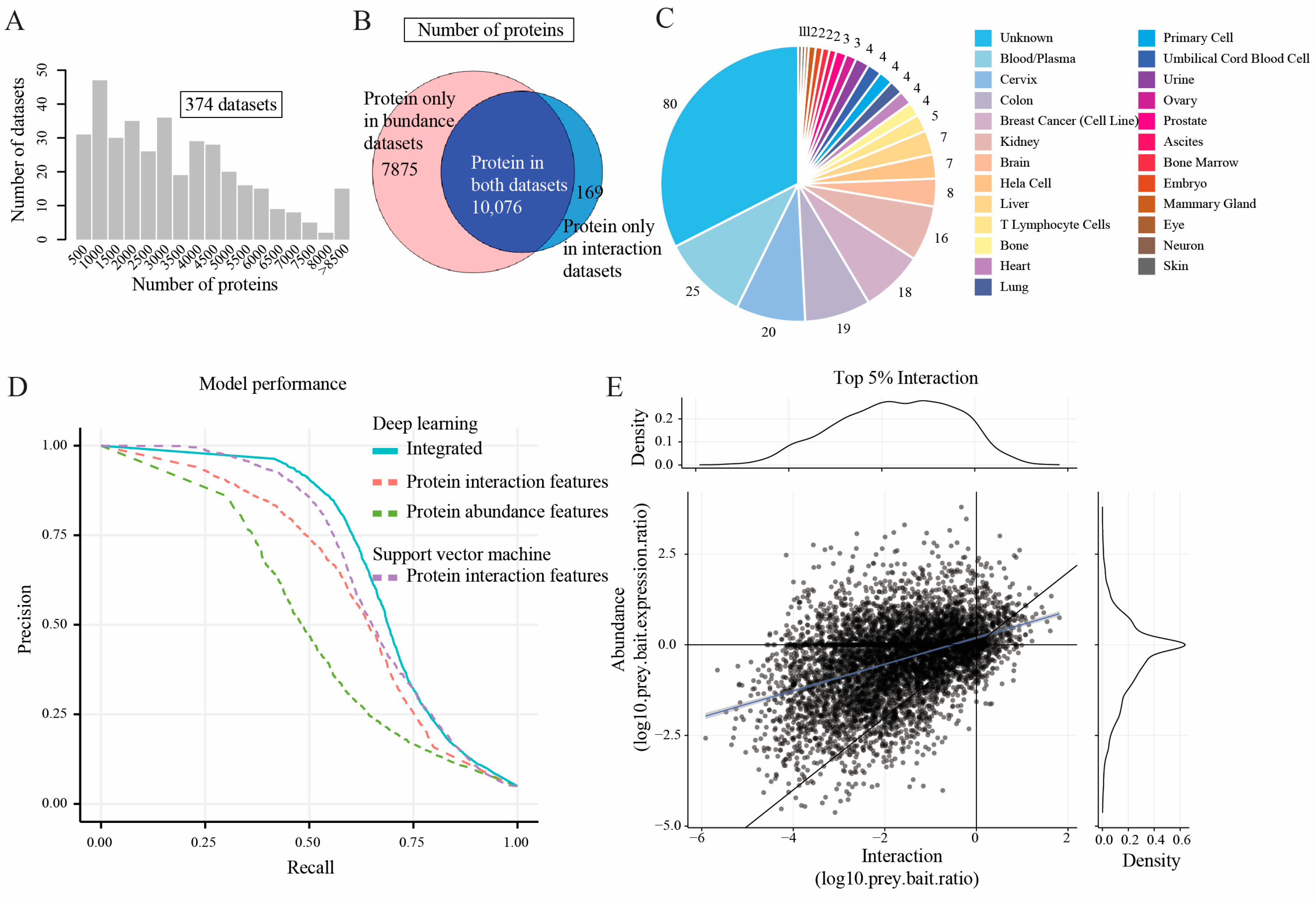
2.1. Feature Matrices Constructed by Incorporating Protein Abundance and Interaction Datasets
2.2. Model Performance Comparison
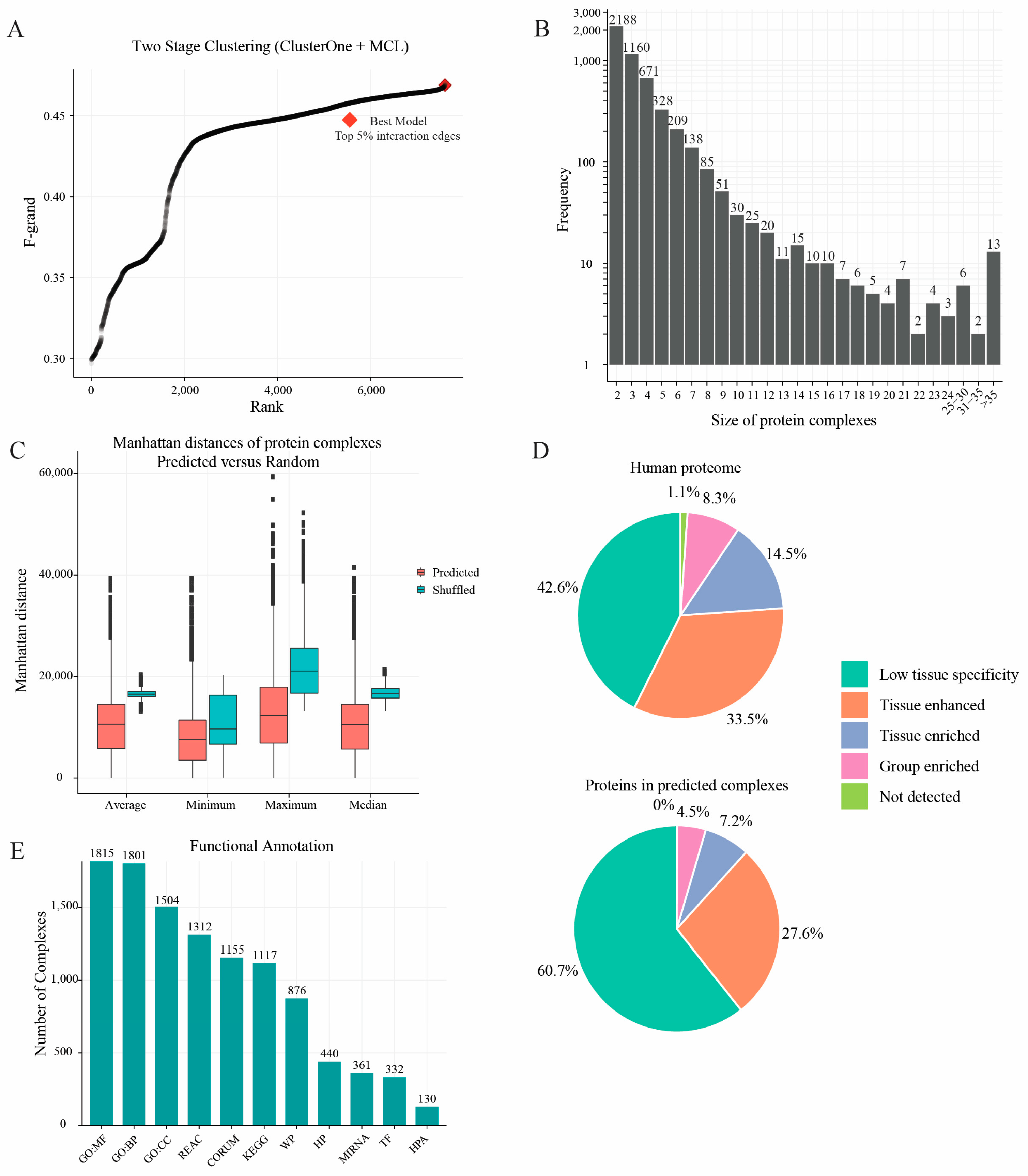
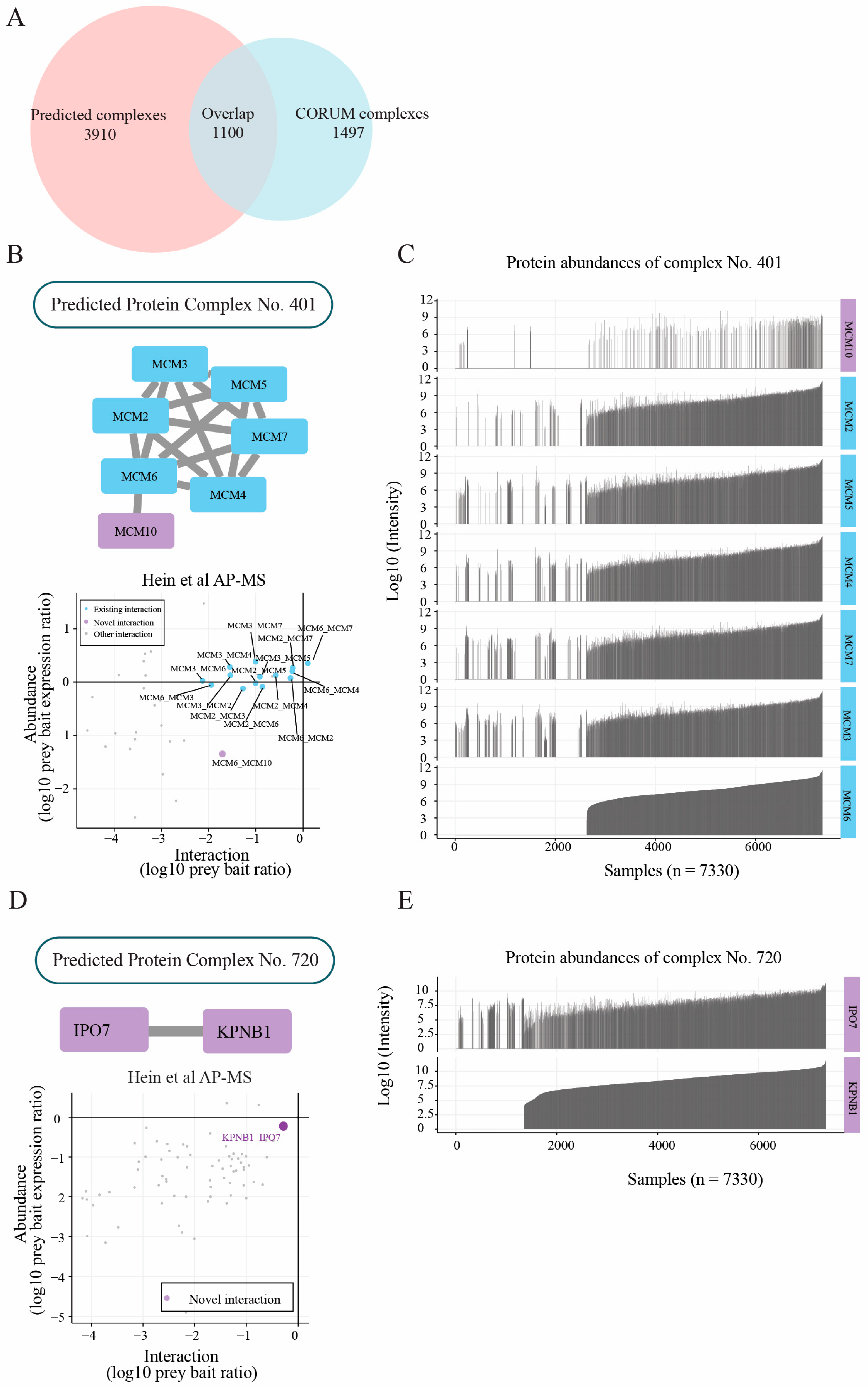
2.3. Protein Complexes Identified by Two-Stage Clustering Method
2.4. Protein Abundance Feature Contributes to Capturing Novel Subunits
2.5. Members of Protein Complexes Exhibit Co-Expression Characteristic
3. Discussion
4. Materials and Methods
4.1. Gold-Standard Reference Set and the Training and Test Protein Pairs
4.2. Featurization of Protein–Protein Interaction Pairs
4.2.1. Protein Abundance Features
4.2.2. Protein Interaction Features
4.3. Deep Learning Neural Network Implementation
4.4. Evaluation of Feature Importance
4.5. Two-Stage Clustering to Predict Protein Complexes
4.6. K-Clique Method-Based Accuracy Evaluation
4.7. Enrichment Analysis and Tissue Specificity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.K.; Dichtl, B. Co-translational control of protein complex formation: A fundamental pathway of cellular organization? Biochem. Soc. Trans. 2018, 46, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem. 2015, 84, 551–575. [Google Scholar] [CrossRef]
- Wu, Z.; Liao, Q.; Liu, B. A comprehensive review and evaluation of computational methods for identifying protein complexes from protein-protein interaction networks. Brief. Bioinform. 2020, 21, 1531–1548. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef]
- Paiano, A.; Margiotta, A.; De Luca, M.; Bucci, C. Yeast Two-Hybrid Assay to Identify Interacting Proteins. Curr. Protoc. Protein Sci. 2019, 95, e70. [Google Scholar] [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Salokas, K.; Tamene, F.; Jiu, Y.; Weldatsadik, R.G.; Ohman, T.; Varjosalo, M. An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 2018, 9, 1188. [Google Scholar] [CrossRef]
- Drew, K.; Muller, C.L.; Bonneau, R.; Marcotte, E.M. Identifying direct contacts between protein complex subunits from their conditional dependence in proteomics datasets. PLoS Comput. Biol. 2017, 13, e1005625. [Google Scholar] [CrossRef]
- Wan, C.H.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.J.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Drew, K.; Lee, C.; Huizar, R.L.; Tu, F.; Borgeson, B.; McWhite, C.D.; Ma, Y.; Wallingford, J.B.; Marcotte, E.M. Integration of over 9000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017, 13, 932. [Google Scholar] [CrossRef]
- Sarkar, D.; Saha, S. Machine-learning techniques for the prediction of protein-protein interactions. J. Biosci. 2019, 44, 104. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Huttlin, E.L.; Harper, J.W.; Gygi, S.P. BioPlex Display: An Interactive Suite for Large-Scale AP-MS Protein-Protein Interaction Data. J. Proteome Res. 2018, 17, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Zhang, J.X.; Zhong, C.; Huang, Y.R.; Lin, H.X.; Wang, M. A method for identifying protein complexes with the features of joint co-localization and joint co-expression in static PPI networks. Comput. Biol. Med. 2019, 111, 103333. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.W.; Minguez, P.; Bork, P.; Auburger, J.J.; Guilbride, D.L.; Kramer, G.; Bukau, B. Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science 2015, 350, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.D.; Liu, F.; Luo, S.Y.; Yin, X.H.; He, D.Q.; Liu, J.G.; Yue, Z.H.; Song, J.K. Co-expression of key gene modules and pathways of human breast cancer cell lines. Biosci. Rep. 2019, 39, Bsr20181925. [Google Scholar] [CrossRef]
- Liu, J.H.; Zhou, S.L.; Li, S.Y.; Jiang, Y.; Wan, Y.C.; Ma, X.L.; Cheng, W.J. Eleven genes associated with progression and prognosis of endometrial cancer (EC) identified by comprehensive bioinformatics analysis. Cancer Cell. Int. 2019, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Rost, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying protein interaction dynamics by SWATH mass spectrometry: Application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456, Erratum in Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. CORUM: The comprehensive resource of mammalian protein complexes-2009. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef]
- Chen, H.L.; Zhou, H.X. Prediction of interface residues in protein-protein complexes by a consensus neural network method: Test against NMR data. Proteins-Struct. Funct. Bioinform. 2005, 61, 21–35. [Google Scholar] [CrossRef]
- Lage, K.; Karlberg, E.O.; Storling, Z.M.; Olason, P.I.; Pedersen, A.G.; Rigina, O.; Hinsby, A.M.; Tumer, Z.; Pociot, F.; Tommerup, N.; et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007, 25, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Nepusz, T.; Yu, H.Y.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, U471–U481. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Kopp, F.; Dahlmann, B.; Kuehn, L. Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28 alpha beta activator: Ultrastructure and peptidase activities. J. Mol. Biol. 2001, 313, 465–471. [Google Scholar] [CrossRef]
- Sato, S.; Tomomori-Sato, C.; Parmely, T.J.; Florens, L.; Zybailov, B.; Swanson, S.K.; Banks, C.A.S.; Jin, J.J.; Cai, Y.; Washburn, M.P.; et al. A set of consensus mammalian Mediator subunits identified by multidimensional protein identification technology. Mol. Cell 2004, 14, 685–691. [Google Scholar] [CrossRef]
- Homesley, L.; Lei, M.; Kawasaki, Y.; Sawyer, S.; Christensen, T.; Tye, B.K. Mcm10 and the MCM2-7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes. Dev. 2000, 14, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Diffley, J.F.X. Recruitment of Mcm10 to Sites of Replication Initiation Requires Direct Binding to the Minichromosome Maintenance (MCM) Complex. J. Biol. Chem. 2016, 291, 5879–5888. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef]
- Jakel, S.; Gorlich, D. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 1998, 17, 4491–4502. [Google Scholar] [CrossRef]
- Jakel, S.; Albig, W.; Kutay, U.; Bischoff, F.R.; Schwamborn, K.; Doenecke, D.; Gorlich, D. The importin beta/importin 7 heterodimer is a functional nuclear import receptor for histone H1. EMBO J. 1999, 18, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, C.L.; Warrington, J.A.; Treadwell, L.; Norcum, M.T. A three-dimensional working model of the multienzyme complex of aminoacyl-tRNA synthetases based on electron microscopic placements of tRNA and proteins. J. Biol. Chem. 2005, 280, 38870–38878. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Granovetter, M.S. The strength of weak ties. Am. J. Sociol. 1973, 78, 1360–1380. [Google Scholar] [CrossRef]
- Csermely, P. Weak Links: Stabilizers of Complex Systems from Proteins to Social Networks; Springer: Berlin/Heidelberg, Germany, 2006; p. 37. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome—History, character, and diagnostic prospects. Mol. Cell Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Petrey, D.; Zhao, H.; Trudeau, S.J.; Murray, D.; Honig, B. PrePPI: A Structure Informed Proteome-wide Database of Protein-Protein Interactions. J. Mol. Biol. 2023, 168052. [Google Scholar] [CrossRef]
- Important facts about cancer. Boston Med. Surg. J. 1920, 182, 125–126.
- Hu.Map Database. Available online: http://hu1.proteincomplexes.org/download (accessed on 10 May 2018).
- Lee, I.; Blom, U.M.; Wang, P.I.; Shim, J.E.; Marcotte, E.M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011, 21, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Guruharsha, K.G.; Rual, J.F.; Zhai, B.; Mintseris, J.; Vaidya, P.; Vaidya, N.; Beekman, C.; Wong, C.; Rhee, D.Y.; Cenaj, O.; et al. A Protein Complex Network of Drosophila melanogaster. Cell 2011, 147, 690–703. [Google Scholar] [CrossRef]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the Human Endogenous Coregulator Complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef]
- R Interface of Keras. Available online: https://keras.rstudio.com (accessed on 10 March 2018).
- Tieleman, T.; Hinton, G. Lecture 6.5-rmsprop: Divide the gradient by a running average of its recent magnitude. COURSERA: Neural Netw. Mach. Learn. 2012, 4, 26–31. [Google Scholar]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F.; Chang, C.; Lin, C. e1071: Misc Functions of the Department of Statistics (e1071), R Package Version 1.7.2; TU Wien: Vienna, Austria, 2014. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: https://www.proteinatlas.org/about/download,proteinatlas.tsv.zip (accessed on 15 May 2019).
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, M.; Klymenko, T.; Fraterman, S.; Papp, B.; Oktaba, K.; Kocher, T.; Cohen, A.; Stunnenberg, H.G.; Wilm, M.; Muller, J. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J. 2007, 26, 4078–4088. [Google Scholar] [CrossRef]
- Mishima, M.; Kaitna, S.; Glotzer, M. Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev. Cell 2002, 2, 41–54. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Wu, B.; Chu, X.Y.; Feng, C.; Hou, J.W.; Fan, H.X.; Liu, N.N.; Li, C.F.; Kong, X.P.; Ye, X.; Meng, S.D. Heat shock protein gp96 decreases p53 stability by regulating Mdm2 E3 ligase activity in liver cancer. Cancer Lett. 2015, 359, 325–334. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Altelaar, M.; van Breukelen, B. Identification of Protein Complexes by Integrating Protein Abundance and Interaction Features Using a Deep Learning Strategy. Int. J. Mol. Sci. 2023, 24, 7884. https://doi.org/10.3390/ijms24097884
Li B, Altelaar M, van Breukelen B. Identification of Protein Complexes by Integrating Protein Abundance and Interaction Features Using a Deep Learning Strategy. International Journal of Molecular Sciences. 2023; 24(9):7884. https://doi.org/10.3390/ijms24097884
Chicago/Turabian StyleLi, Bohui, Maarten Altelaar, and Bas van Breukelen. 2023. "Identification of Protein Complexes by Integrating Protein Abundance and Interaction Features Using a Deep Learning Strategy" International Journal of Molecular Sciences 24, no. 9: 7884. https://doi.org/10.3390/ijms24097884
APA StyleLi, B., Altelaar, M., & van Breukelen, B. (2023). Identification of Protein Complexes by Integrating Protein Abundance and Interaction Features Using a Deep Learning Strategy. International Journal of Molecular Sciences, 24(9), 7884. https://doi.org/10.3390/ijms24097884