

APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer

 ,
,  ,
,
Abstract
1. Introduction
2. Results
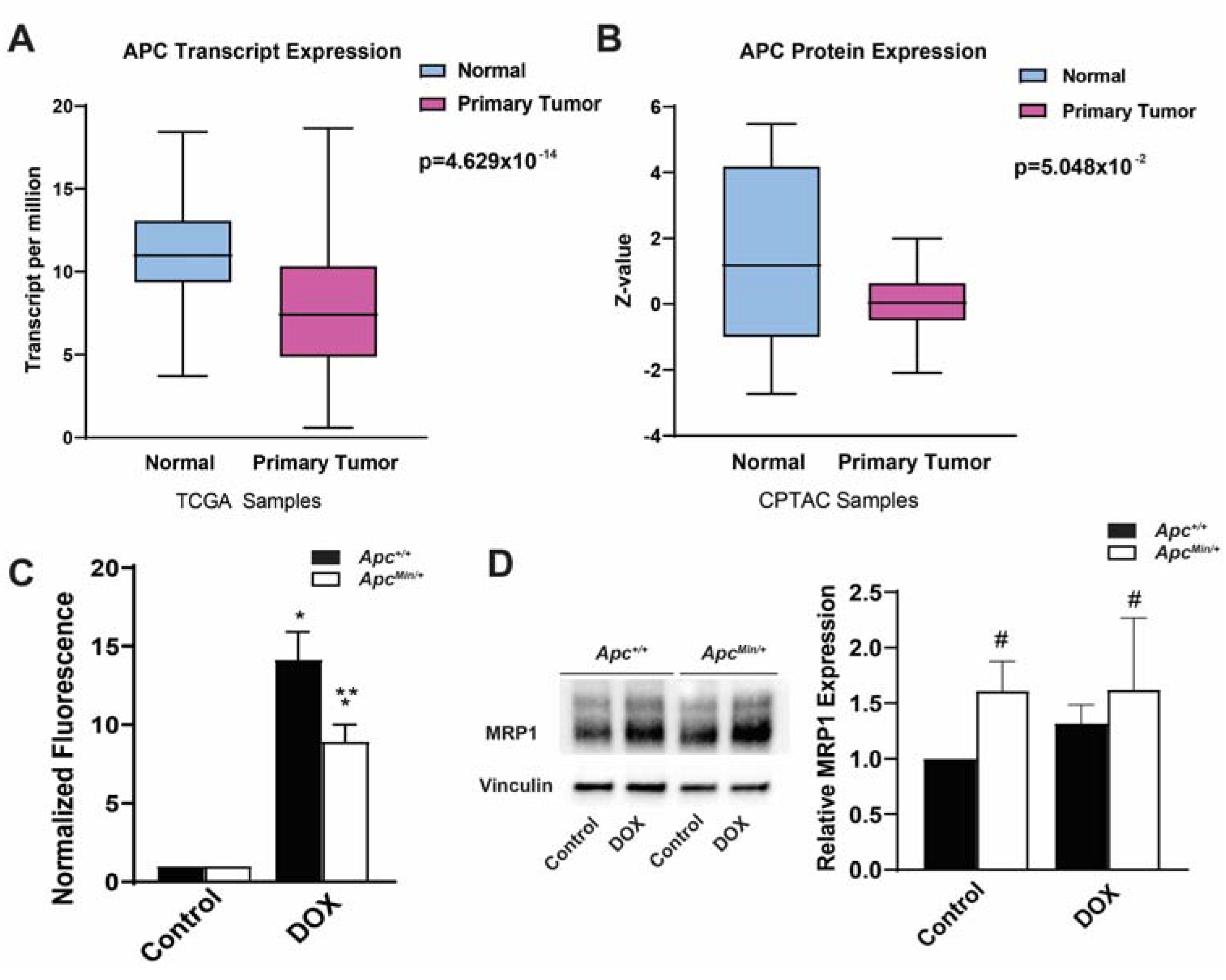
2.1. Decreased APC Expression in Breast Cancer
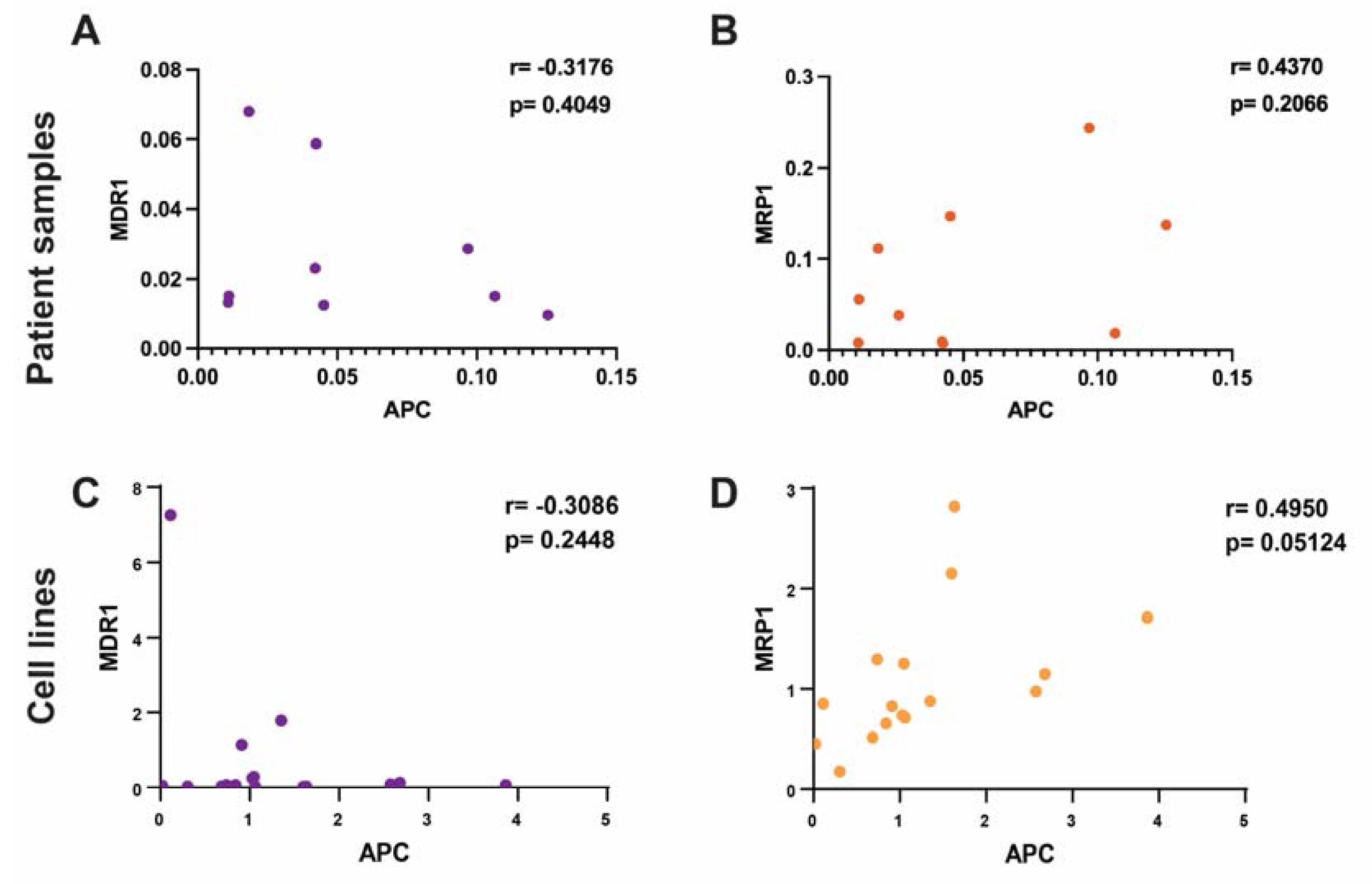
2.2. Correlation between APC Expression Levels and ABC Transporters
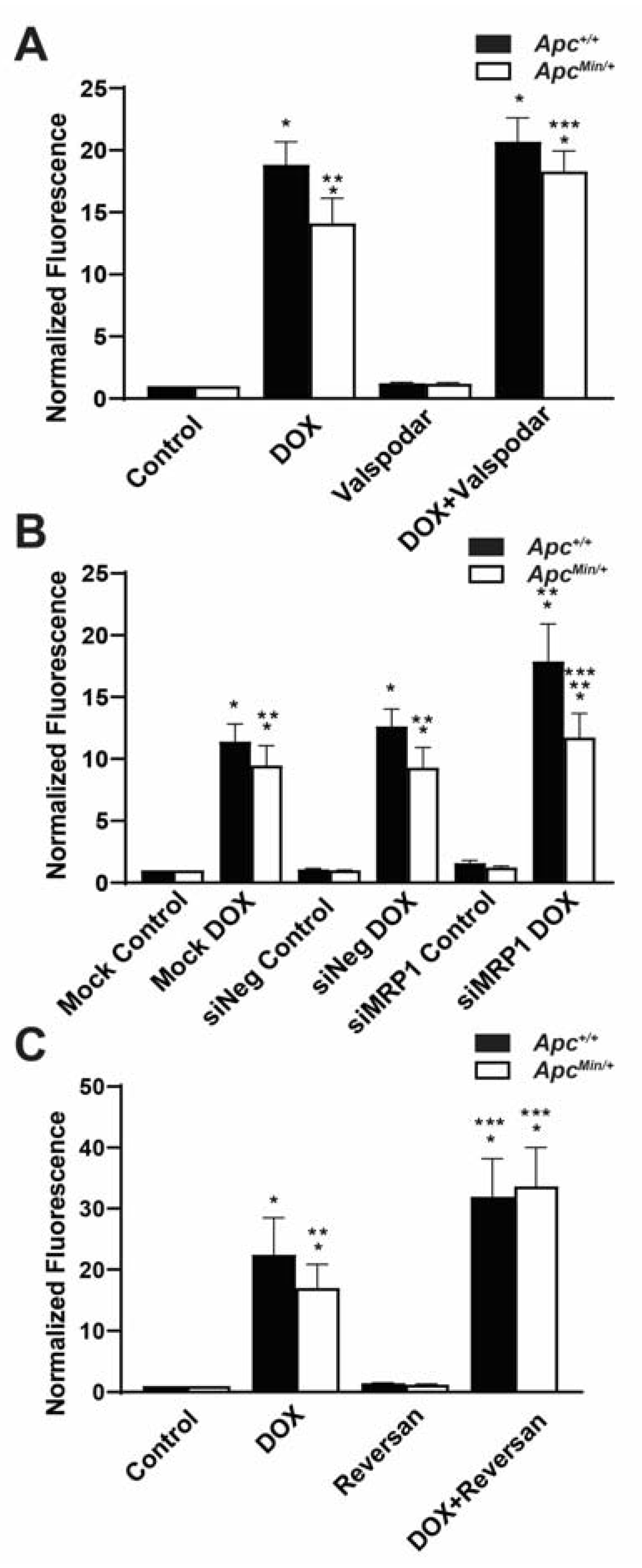
2.3. MDR1 and MRP1 Activity Caused Decreased Intracellular DOX Accumulation
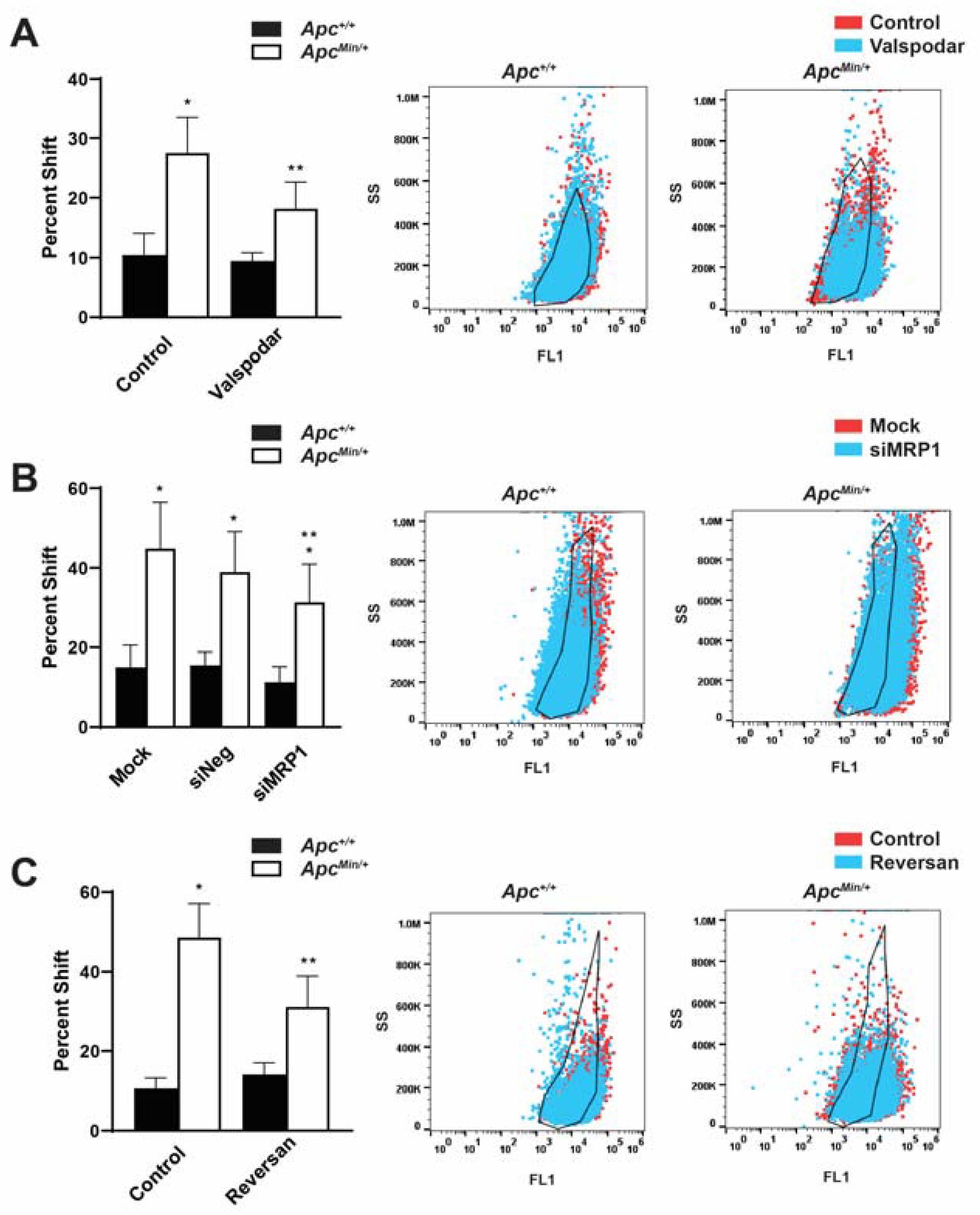
2.4. Inhibition of MDR1 and MRP1 Alone and in Combination Decreased the Population of TIC as Measured by ALDHhigh Cell Populations
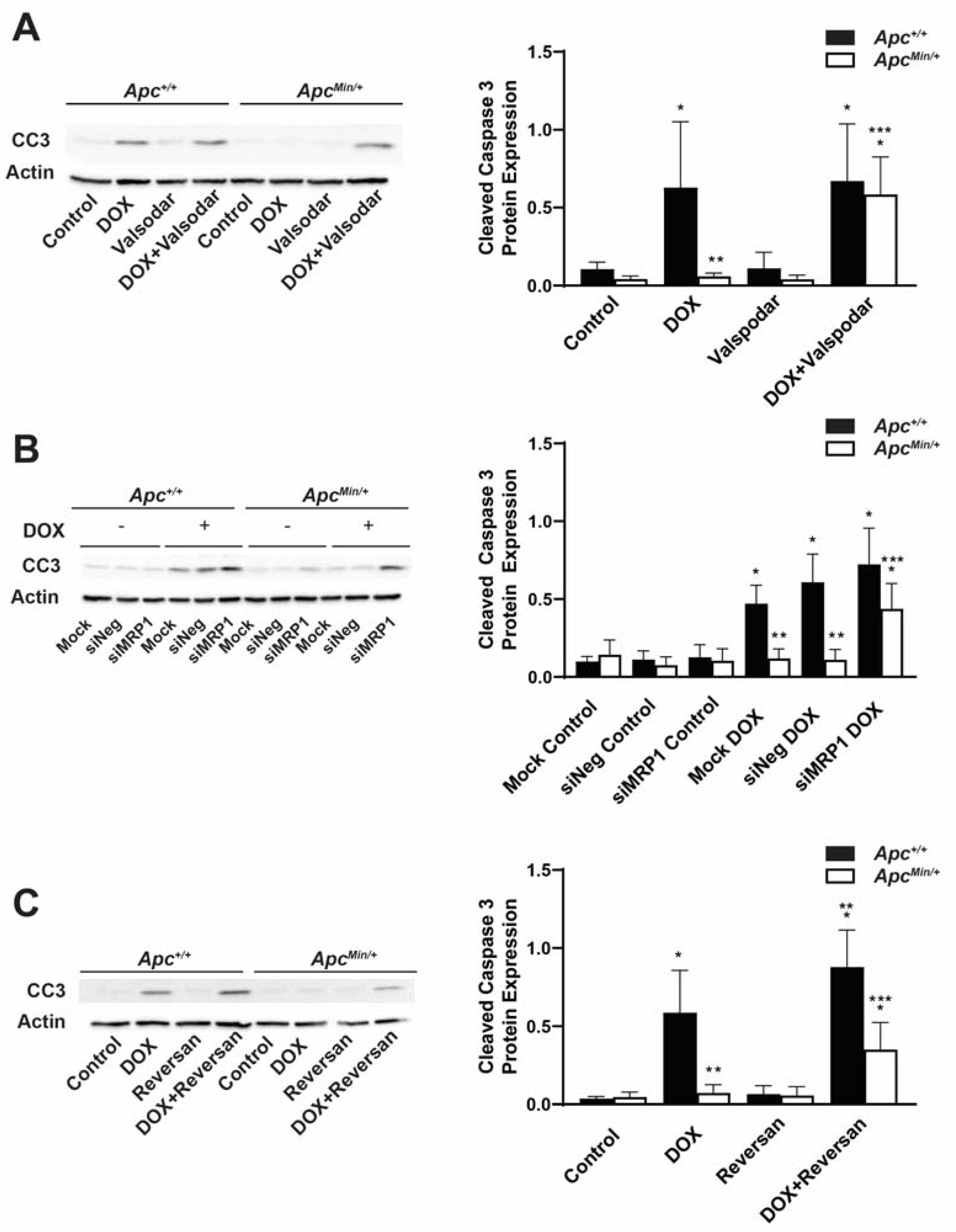
2.5. Increased ABC Transporters Cause DOX Resistance
3. Discussion
4. Materials and Methods
4.1. Database Analysis
4.2. Cell Culture
4.3. Patient Samples
4.4. Drug Treatment
4.5. Cell Viability
4.6. SiRNA Transfection
4.7. Western Blotting
4.8. Aldefluor Assay
4.9. Measurement of Intracellular DOX
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ABC | Adenosine-binding cassette |
| APC | Adenomatous Polyposis Coli |
| ALDH | Aldehyde dehydrogenase |
| BCRP/ABCG2 | Breast cancer resistance protein |
| CC3 | Cleaved caspase 3 |
| DOX | Doxorubicin |
| ER | Estrogen receptor |
| HER2 | Human epidermal growth factor |
| lncRNA | Long noncoding RNA |
| MDR | Multidrug resistance |
| MRP1/ABBC1 | Multidrug resistance-associated protein 1 |
| MRD1/ABCB1 | Multidrug resistance protein 1 |
| P/S | Penicillin/streptomycin |
| PyMT | Polyoma middle T antigen |
| PR | Progesterone receptor |
| STAT3 | Signal transducer and activation of transcription 3 |
| TNBC | Triple-negative breast cancer |
| TPBC | Triple-positive breast cancer |
References
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple Negative Breast Cancer—An Overview. Hered. Genet. 2013, 2013 (Suppl. S2). [Google Scholar] [CrossRef]
- Prasad, C.P.; Mirza, S.; Sharma, G.; Prashad, R.; DattaGupta, S.; Rath, G.; Ralhan, R. Epigenetic alterations of CDH1 and APC genes: Relationship with activation of Wnt/beta-catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008, 83, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Bhattacharya, N.; Alam, N.; Roy, A.; Roychoudhury, S.; Panda, C.K. Subtype-specific alterations of the Wnt signaling pathway in breast cancer: Clinical and prognostic significance. Cancer Sci. 2012, 103, 210–220. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Van Laere, S.J.; Van den Bosch, S.M.; Van den Eynden, G.G.; Trinh, B.X.; van Dam, P.A.; Colpaert, C.G.; van Engeland, M.; Van Marck, E.A.; Vermeulen, P.B.; et al. Aberrant methylation of the Adenomatous Polyposis Coli (APC) gene promoter is associated with the inflammatory breast cancer phenotype. Br. J. Cancer 2008, 99, 1735–1742. [Google Scholar] [CrossRef]
- VanKlompenberg, M.K.; Bedalov, C.O.; Soto, K.F.; Prosperi, J.R. APC selectively mediates response to chemotherapeutic agents in breast cancer. BMC Cancer 2015, 15, 457. [Google Scholar] [CrossRef]
- Prosperi, J.R.; Khramtsov, A.I.; Khramtsova, G.F.; Goss, K.H. Apc mutation enhances PyMT-induced mammary tumorigenesis. PLoS ONE 2011, 6, e29339. [Google Scholar] [CrossRef]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Perez, E.A. Impact, mechanisms, and novel chemotherapy strategies for overcoming resistance to anthracyclines and taxanes in metastatic breast cancer. Breast Cancer Res. Treat. 2008, 114, 195. [Google Scholar] [CrossRef]
- Manciu, L.; Chang, X.-B.; Buyse, F.; Hou, Y.-X.; Gustot, A.; Riordan, J.R.; Ruysschaert, J.M. Intermediate Structural States Involved in MRP1-Mediated Drug Transport: ROLE OF GLUTATHIONE. J. Biol. Chem. 2003, 278, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: Future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist. 2021, 4, 784–804. [Google Scholar] [CrossRef] [PubMed]
- VanKlompenberg, M.K.; Leyden, E.; Arnason, A.H.; Zhang, J.T.; Stefanski, C.D.; Prosperi, J.R. APC loss in breast cancer leads to doxorubicin resistance via STAT3 activation. Oncotarget 2017, 8, 102868–102879. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Lorendeau, D.; Khonkarn, R.; Dury, L.; Pérès, B.; Boumendjel, A.; Cortay, J.-C.; Falson, P.; Chaptal, V.; Baubichon-Cortay, H. Molecular analysis of the massive GSH transport mechanism mediated by the human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 2020, 10, 7616. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and Its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Tanić, N.; Prvanović, M.; Milovanović, Z.; Tanić, N. Friend or foe: ABCG2, ABCC1 and ABCB1 expression in triple-negative breast cancer. Breast Cancer 2021, 28, 727–736. [Google Scholar] [CrossRef]
- Zheng, Y.; Ma, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar]
- Iorio, M.V.; Croce, C.M. MicroRNAs in cancer: Small molecules with a huge impact. J. Clin. Oncol. 2009, 27, 5848–5856. [Google Scholar] [CrossRef]
- Chen, J.; Tian, W.; Cai, H.; He, H.; Deng, Y. Down-regulation of microRNA-200c is associated with drug resistance in human breast cancer. Med. Oncol. 2012, 29, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Zangouei, A.S.; Alimardani, M.; Moghbeli, M. MicroRNAs as the critical regulators of Doxorubicin resistance in breast tumor cells. Cancer Cell Int. 2021, 21, 213. [Google Scholar] [CrossRef]
- Lu, L.; Ju, F.; Zhao, H.; Ma, X. MicroRNA-134 modulates resistance to doxorubicin in human breast cancer cells by downregulating ABCC1. Biotechnol. Lett. 2015, 37, 2387–2394. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, W.; Yuan, Z.; Liu, X.; Jiang, H. LncRNA-MALAT1 contributes to the cisplatin-resistance of lung cancer by upregulating MRP1 and MDR1 via STAT3 activation. Biomed. Pharmacother. 2018, 101, 536–542. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Crocker, P.R.; Eklund, R.C.; Graham, T.R. Evaluating the factorial structure of the revised causal dimension scale in adolescents. Res. Q. Exerc. Sport 2002, 73, 211–218. [Google Scholar] [CrossRef]
- Kumar, B.; Prasad, M.; Bhat-Nakshatri, P.; Anjanappa, M.; Kalra, M.; Marino, N.; Storniolo, A.M.; Rao, X.; Liu, S.; Wan, J.; et al. Normal Breast-Derived Epithelial Cells with Luminal and Intrinsic Subtype-Enriched Gene Expression Document Interindividual Differences in Their Differentiation Cascade. Cancer Res. 2018, 78, 5107–5123. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.J.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G.; et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016, 35, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.-Y.; Zhang, J.-T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Dong, Z.; Qi, J.; Yang, Y.; Liu, Y.; Li, Z.; Xu, J.; Zhang, J.-T. A Novel Two Mode-Acting Inhibitor of ABCG2-Mediated Multidrug Transport and Resistance in Cancer Chemotherapy. PLoS ONE 2009, 4, e5676. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ER | PR | HER2 |
|---|---|---|---|
| ND978 | + | − | − |
| ND1063 | + | + | − |
| ND1114 | + | + | − |
| ND1116 | + | + | + |
| ND1167 | + | + | − |
| ND1195 | + | + | − |
| ND1205 | + | + | − |
| ND1210 | − | − | − |
| ND1222 | + | + | − |
| ND1230 | + | + | − |
| ND1240 | + | + | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefanski, C.D.; Arnason, A.; Maloney, S.; Kotsen, J.; Powers, E.; Zhang, J.-T.; Prosperi, J.R. APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 7621. https://doi.org/10.3390/ijms24087621
Stefanski CD, Arnason A, Maloney S, Kotsen J, Powers E, Zhang J-T, Prosperi JR. APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer. International Journal of Molecular Sciences. 2023; 24(8):7621. https://doi.org/10.3390/ijms24087621
Chicago/Turabian StyleStefanski, Casey D., Anne Arnason, Sara Maloney, Janna Kotsen, Elizabeth Powers, Jian-Ting Zhang, and Jenifer R. Prosperi. 2023. "APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer" International Journal of Molecular Sciences 24, no. 8: 7621. https://doi.org/10.3390/ijms24087621
APA StyleStefanski, C. D., Arnason, A., Maloney, S., Kotsen, J., Powers, E., Zhang, J.-T., & Prosperi, J. R. (2023). APC Loss Prevents Doxorubicin-Induced Cell Death by Increasing Drug Efflux and a Chemoresistant Cell Population in Breast Cancer. International Journal of Molecular Sciences, 24(8), 7621. https://doi.org/10.3390/ijms24087621