
HIV Promotes Atherosclerosis via Circulating Extracellular Vesicle MicroRNAs

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PLHIV Have Increased Carotid Intima-Media Thickness (cIMT) and Decreased ECFC Numbers
2.2. HIVposEVs Promote Atherogenesis in apoE−/− Mice
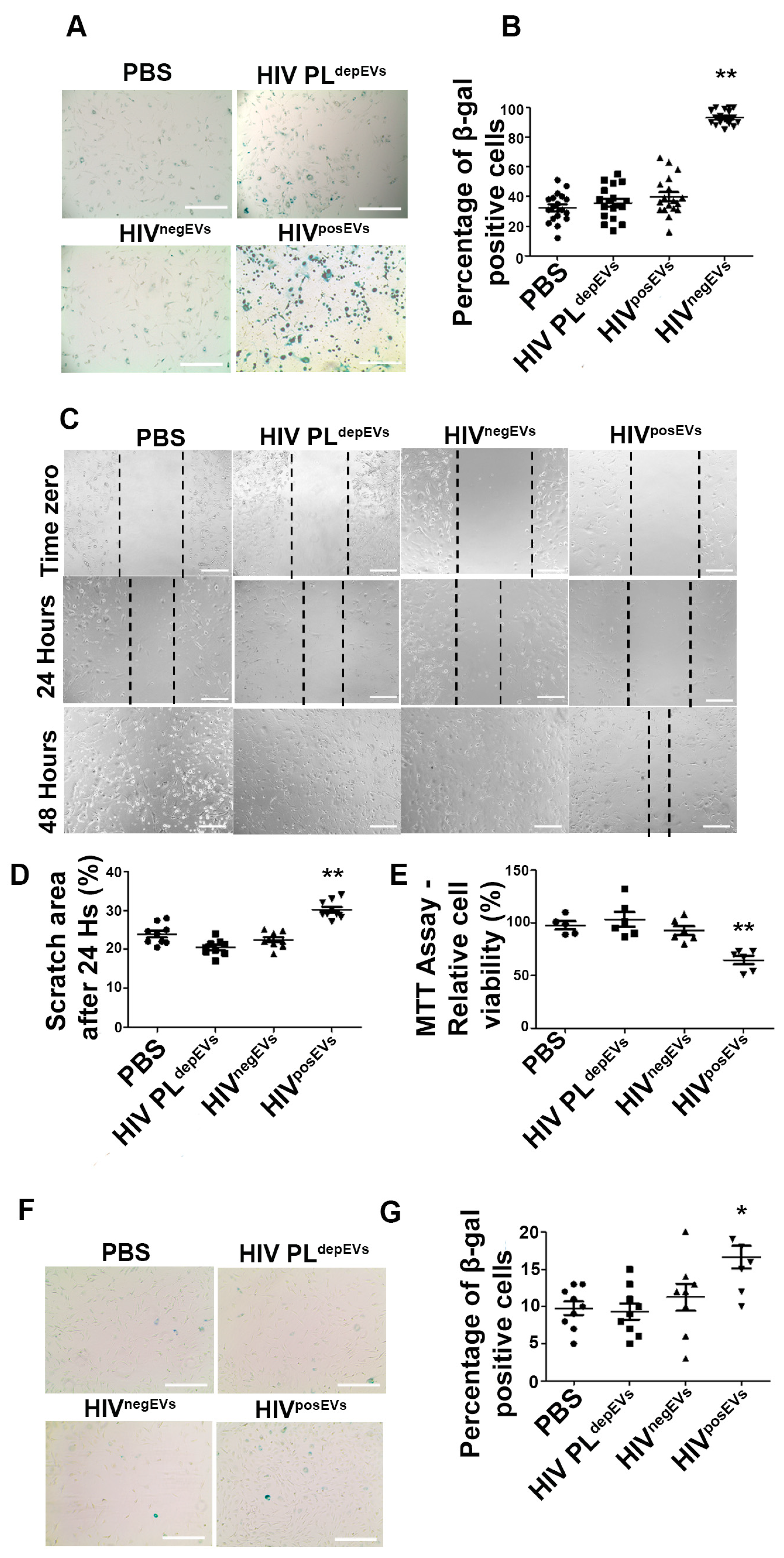
2.3. HIVposEVs Cause Vascular Cell Senescence and Apoptosis In Vivo
2.4. HIVposEVs Affect lin− BMC Senescence and Functionality
2.5. HIVposEVs Impairs EC Functionality
2.6. HIVposEVs and HIVnegEVs Show Similar Physical Characteristics and Surface Markers
2.7. HIVposEVs Have Differential microRNA Cargo Compared to HIVnegEVs
2.8. TEVs Overexpressing Antagomirs for Candidate EV-miRs Attenuate the HIVposEVs Effects
2.9. Overexpression of Hmga2 Rescues the Effects of HIVposEVs on lin− BMCs
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Isolation & Characterization of Plasma EVs
4.3. EVs with Modified miR Contents
4.4. Animal Model
4.5. Atherosclerosis Measurement
4.6. Quantification of Endothelial Colony-Forming Cells (ECFCs) from Humans
4.7. Isolation of lin− BMCs from Mice
4.8. Lipid Panel and C Reactive Protein ELISA Assays
4.9. Cell Migration “Scratch” Assay
4.10. Apoptosis Assay
4.11. Senescence Assay
4.12. Cell Proliferation Assay
4.13. Small RNA Sequencing (sRNA-seq)
4.14. qRT-PCR Validation of Candidate EV-miRs
4.15. Western Blot
4.16. Ingenuity Pathway Analyses (IPA)
4.17. Transduction of Mouse lin− BMCs
4.18. In Vitro EV Treatment
4.19. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ART | Antiretroviral therapy |
| BM | Bone marrow |
| CFU | Colony forming units |
| cIMT | Carotid intima-media thickness |
| CVD | Cerebrocardiovascular disease |
| ECs | Endothelial cells |
| ECFCs | Endothelial colony-forming cells |
| EPCs | Endothelial progenitor cells |
| EV-miRs | extracellular vesicles microRNAs |
| EVs | Extracellular vesicles |
| FDR | False discovery rate |
| HIVnegEVs | Extracellular vesicles from HIV negative plasma |
| HIVposEVs | Extracellular vesicles from PLHIV plasma |
| HIV PLdepEVs | HIV plasma depleted of EVs |
| IACUC | Institutional Animal Care and Use Committee |
| IPA | Ingenuity Pathway Analysis |
| Lin− BMCs | Lineage negative bone marrow cells |
| miRs | microRNAs |
| MSC | mesenchymal stromal cell |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide |
| NTA | Nanoparticle Tracking Analysis |
| ORO | Oil Red O |
| PLHIV | People Living with HIV |
| SMCs | Smooth muscle cells |
| sRNA-Seq | small RNA Sequencing |
| TEVs | Tailored extracellular vesicles |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling |
| β-gal | β-galactosidase |
References
- Fauci, A.S.; Lane, H.C. Four Decades of HIV/AIDS—Much Accomplished, Much to Do. N. Engl. J. Med. 2020, 383, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Currier, J.S.; Lundgren, J.; Carr, A.; Klein, D.; Sabin, C.; Sax, P.E.; Schouten, J.T.; Smieja, M. Epidemiological Evidence for Cardiovascular Disease in HIV-Infected Patients and Relationship to Highly Active Antiretroviral Therapy. Circulation 2008, 118, e29–e35. [Google Scholar] [CrossRef]
- Lorenc, A.; Ananthavarathan, P.; Lorigan, J.; Jowata, M.; Brook, G.; Banarsee, R. The prevalence of comorbidities among people living with HIV in Brent: A diverse London Borough. Lond. J. Prim. Care 2014, 6, 84–90. [Google Scholar] [CrossRef]
- Younas, M.; Psomas, C.; Reynes, C.; Cezar, R.; Kundura, L.; Portalès, P.; Merle, C.; Atoui, N.; Fernandez, C.; Le Moing, V.; et al. Residual Viremia Is Linked to a Specific Immune Activation Profile in HIV-1-Infected Adults under Efficient Antiretroviral Therapy. Front. Immunol. 2021, 12, 663843. [Google Scholar] [CrossRef]
- Bhatta, D.N.; Subedi, A.; Sharma, N. Tobacco smoking and alcohol drinking among HIV infected people using antiretroviral therapy. Tob. Induc. Dis. 2018, 16, 16. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Banno, K.; Yoder, M.C. Tissue regeneration using endothelial colony-forming cells: Promising cells for vascular repair. Pediatr. Res. 2018, 83, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Toupance, S.; Simonici, S.; Labat, C.; Dumoulin, C.; Lai, T.; Lakomy, C.; Regnault, V.; Lacolley, P.; George, F.D.; Sabatier, F.; et al. Number and Replating Capacity of Endothelial Colony-Forming Cells are Telomere Length Dependent: Implication for Human Atherogenesis. J. Am. Hear. Assoc. 2021, 10, e020606. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Liu, Z.; Sims, E.C.; Repass, M.J.; Haneline, L.S.; Yoder, M.C. Endothelial colony-forming cell function is reduced during HIV infection. J. Infect. Dis. 2019, 219, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Borghi, C.; Grassi, D.; Cicero, A.F.G. People living with human immunodeficiency virus: Cardiovascular risk screening for an early and effective risk management. Atherosclerosis 2022, 353, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, S. Endothelial Progenitor Cells as Molecular Targets in Vascular Senescence and Repair. Curr. Stem Cell Res. Ther. 2018, 13, 438–446. [Google Scholar] [CrossRef]
- Wang, L.; Wei, J.; Da Fonseca Ferreira, A.; Wang, H.; Zhang, L.; Zhang, Q.; Bellio, M.A.; Chu, X.-M.; Khan, A.; Jayaweera, D.; et al. Rejuvenation of Senescent Endothelial Progenitor Cells by Extracellular Vesicles Derived from Mesenchymal Stromal Cells. JACC Basic Transl. Sci. 2020, 5, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Wang, H.; Jia, C.; Zhu, S.; Chu, X.; Ma, Q.; Wei, J.; Chen, E.; Zhu, W.; Macon, C.J.; et al. MicroRNA-146a Induces Lineage-Negative Bone Marrow Cell Apoptosis and Senescence by Targeting Polo-Like Kinase 2 Expression. Arter. Thromb. Vasc. Biol. 2017, 37, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Ganem, D. MicroRNAs and Viral Infection. Mol. Cell 2005, 20, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Rani, S.; Breslin, S.; Gogarty, M.; Ghobrial, I.M.; Crown, J.; O’driscoll, L. miR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol. Cancer 2014, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Latifkar, A.; Hur, Y.H.; Sanchez, J.C.; Cerione, R.A.; Antonyak, M.A. New insights into extracellular vesicle biogenesis and function. J. Cell Sci. 2019, 132, jcs222406. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef]
- Santangelo, L.; Bordoni, V.; Montaldo, C.; Cimini, E.; Zingoni, A.; Battistelli, C.; D’Offizi, G.; Capobianchi, M.R.; Santoni, A.; Tripodi, M.; et al. Hepatitis C virus direct-acting antivirals therapy impacts on extracellular vesicles microRNAs content and on their immunomodulating properties. Liver Int. 2018, 38, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Ferdin, J.; Goričar, K.; Dolžan, V.; Plemenitaš, A.; Martin, J.N.; Peterlin, B.M.; Deeks, S.G.; Lenassi, M. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS ONE 2018, 13, e0191613. [Google Scholar] [CrossRef] [PubMed]
- Dubrovsky, L.; Brichacek, B.; Prashant, N.M.; Pushkarsky, T.; Mukhamedova, N.; Fleetwood, A.J.; Xu, Y.; Dragoljevic, D.; Fitzgerald, M.; Horvath, A.; et al. Extracellular vesicles carrying HIV-1 Nef induce long-term hyperreactivity of myeloid cells. Cell Rep. 2022, 41, 111674. [Google Scholar] [CrossRef]
- Mukhamedova, N.; Hoang, A.; Dragoljevic, D.; Dubrovsky, L.; Pushkarsky, T.; Low, H.; Ditiatkovski, M.; Fu, Y.; Ohkawa, R.; Meikle, P.J.; et al. Exosomes containing HIV protein Nef reorganize lipid rafts potentiating inflammatory response in bystander cells. PLOS Pathog. 2019, 15, e1007907. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.L.; Guyonnet, L.; Goudot, G.; Revets, D.; Konstantinou, M.; Chipont, A.; Chocron, R.; Blandinieres, A.; Khider, L.; Rancic, J.; et al. Multidimensional Proteomic Approach of Endothelial Progenitors Demonstrate Expression of KDR Restricted to CD19 Cells. Stem Cell Rev. Rep. 2020, 17, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Z.; Davison, F.; Hu, Y. Circulating Progenitor Cells Regenerate Endothelium of Vein Graft Atherosclerosis, Which Is Diminished in ApoE-Deficient Mice. Circ. Res. 2003, 93, e76–e86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Deng, S.; Ma, Q.; Zhang, T.; Jia, C.; Zhuo, D.; Yang, F.; Wei, J.; Wang, L.; Dykxhoorn, D.M.; et al. MicroRNA-10A* and MicroRNA-21 Modulate Endothelial Progenitor Cell Senescence via Suppressing High-Mobility Group A2. Circ. Res. 2013, 112, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Gao, L.; Wu, Z.; Wang, L.; Fan, L. Let-7b-5p promotes cell apoptosis in Parkinson’s disease by targeting HMGA2. Mol. Med. Rep. 2021, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 Promotes Neural Stem Cell Self-Renewal in Young but Not Old Mice by Reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–239. [Google Scholar] [CrossRef]
- Christensen, S.; Wolf, E.; Altevers, J.; Diaz-Cuervo, H. Comorbidities and costs in HIV patients: A retrospective claims database analysis in Germany. PLoS ONE 2019, 14, e0224279. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bezsonov, E.E.; Borisov, E.E.; Grechko, A.V.; Kartuesov, A.G.; Orekhov, A.N. Atherosclerosis in HIV patients: What do we know so far? Int. J. Mol. Sci. 2022, 23, 2504. [Google Scholar] [CrossRef]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef]
- Kulkarni, M.; Bowman, E.; Gabriel, J.; Amburgy, T.; Mayne, E.; Zidar, D.A.; Maierhofer, C.; Turner, A.N.; Bazan, J.A.; Koletar, S.L.; et al. Altered Monocyte and Endothelial Cell Adhesion Molecule Expression Is Linked to Vascular Inflammation in Human Immunodeficiency Virus Infection. Open Forum Infect. Dis. 2016, 3, ofw224. [Google Scholar] [CrossRef]
- Feinstein, M.J.; Bogorodskaya, M.; Bloomfield, G.S.; Vedanthan, R.; Siedner, M.J.; Kwan, G.F.; Longenecker, C.T. Cardiovascular Complications of HIV in Endemic Countries. Curr. Cardiol. Rep. 2016, 18, 113. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Atkinson, J.B.; Fazio, S. Prevention of Atherosclerosis in Apolipoprotein E-Deficient Mice by Bone Marrow Transplantation. Science 1995, 267, 1034–1037. [Google Scholar] [CrossRef]
- Papasavvas, E.; Hsue, P.; Reynolds, G.; Pistilli, M.; Hancock, A.; Martin, J.N.; Deeks, S.G.; Montaner, L.J. Increased CD34+/KDR+ cells are not associated with carotid artery intima-media thickness progression in chronic HIV-positive subjects. Antivir. Ther. 2012, 17, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1–Associated Atherosclerosis: Unraveling the Missing Link. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Zhao, C.; Sun, G.; Li, S.; Lang, M.-F.; Yang, S.; Li, W.; Shi, Y. MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 1876–1881. [Google Scholar] [CrossRef]
- Long, G.; Wang, F.; Duan, Q.; Yang, S.; Chen, F.; Gong, W.; Yang, X.; Wang, Y.; Chen, C.; Wang, D.W. Circulating miR-30a, miR-195 and let-7b Associated with Acute Myocardial Infarction. PLoS ONE 2012, 7, e50926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Su, H.; Ma, X.; Xu, X.; Liang, L.; Ma, G.; Shi, L. MiRNA let-7b promotes the development of hypoxic pulmonary hypertension by targeting ACE2. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L547–L557. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.K.; Kim, C.-H.; Jung, H.Y.; Lee, D.R.; Kim, J.K. Discovery and characterization of miRNA during cellular senescence in bone marrow-derived human mesenchymal stem cells. Exp. Gerontol. 2014, 58, 139–145. [Google Scholar] [CrossRef]
- Lu, Q.; Ma, Z.; Ding, Y.; Bedarida, T.; Chen, L.; Xie, Z.; Song, P.; Zou, M.-H. Circulating miR-103a-3p contributes to angiotensin II-induced renal inflammation and fibrosis via a SNRK/NF-κB/p65 regulatory axis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Okamoto, M.; Fukushima, Y.; Kouwaki, T.; Daito, T.; Kohara, M.; Kida, H.; Oshiumi, H. MicroRNA-451a in extracellular, blood-resident vesicles attenuates macrophage and dendritic cell responses to influenza whole-virus vaccine. J. Biol. Chem. 2018, 293, 18585–18600. [Google Scholar] [CrossRef]
- Tzatsos, A.; Bardeesy, N. Ink4a/Arf Regulation by let-7b and Hmga2: A Genetic Pathway Governing Stem Cell Aging. Cell Stem Cell 2008, 3, 469–470. [Google Scholar] [CrossRef]
- Hammond, S.M.; Sharpless, N.E. HMGA2, MicroRNAs, and Stem Cell Aging. Cell 2008, 135, 1013–1016. [Google Scholar] [CrossRef]
- Nishino, J.; Kim, S.; Zhu, Y.; Zhu, H.; Morrison, S.J. A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. Elife 2013, 2, e00924. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Gray, W.D.; Hayek, S.S.; Ko, Y.-A.; Thomas, S.; Rooney, K.; Awad, M.; Roback, J.D.; Quyyumi, A.; Searles, C.D. Platelets confound the measurement of extracellular miRNA in archived plasma. Sci. Rep. 2016, 6, 32651. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, E.G.M.; Deng, X.; Liu, J.; Bowler, S.A.; Shikuma, C.M.; Stone, M.; Hunt, P.W.; Ndhlovu, L.C.; Norris, P.J. Plasma CD16+ Extracellular Vesicles Associate with Carotid Artery Intima-Media Thickness in HIV+ Adults on Combination Antiretroviral Therapy. Mbio 2022, 13, 139–145. [Google Scholar] [CrossRef]
- Chelvanambi, S.; Bogatcheva, N.; Bednorz, M.; Agarwal, S.; Maier, B.; Alves, N.J.; Li, W.; Syed, F.; Saber, M.M.; Dahl, N.; et al. HIV-Nef Protein Persists in the Lungs of Aviremic Patients with HIV and Induces Endothelial Cell Death. Am. J. Respir. Cell Mol. Biol. 2019, 60, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Rundek, T.; Elkind, M.S.; Pittman, J.; Boden-Albala, B.; Martin, S.; Humphries, S.E.; Juo, S.H.; Sacco, R.L. Carotid Intima-Media Thickness Is Associated with Allelic Variants of Stromelysin-1, Interleukin-6, and Hepatic Lipase Genes. Stroke 2002, 33, 1420–1423. [Google Scholar] [CrossRef]
- Touboul, P.J.; Hennerici, M.; Meairs, S.; Adams, H.; Amarenco, P.; Desvarieux, M.; Ebrahim, S.; Fatar, M.; Hernandez, R.H.; Kownator, S.; et al. Mannheim Intima-Media Thickness Consensus. Cerebrovasc. Dis. 2004, 18, 346–349. [Google Scholar] [CrossRef]
- Sacco, R.L.; Anand, K.; Lee, H.S.; Boden-Albala, B.; Stabler, S.; Allen, R.; Paik, M.C. Homocysteine and the risk of ischemic stroke in a triethnic cohort: The NOrthern MAnhattan Study. Stroke 2004, 35, 2263–2269. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Chapter 3: Isolation and characterization of exosomes from cell culture supernatants and biological fluids. In Current Protocols in Cell Biology; John Wiley&Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Osteikoetxea, X.; Balogh, A.; Szabó-Taylor, K.; Németh, A.; Szabó, T.G.; Pálóczi, K.; Sódar, B.; Kittel, Á.; György, B.; Pállinger, É.; et al. Improved Characterization of EV Preparations Based on Protein to Lipid Ratio and Lipid Properties. PLoS ONE 2015, 10, e0121184. [Google Scholar] [CrossRef]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011.
- Matveeva, V.; Khanova, M.; Sardin, E.; Antonova, L.; Barbarash, O. Endovascular Interventions Permit Isolation of Endothelial Colony-Forming Cells from Peripheral Blood. Int. J. Mol. Sci. 2018, 19, 3453. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Peterson, M.F.; Otoc, N.; Sethi, J.K.; Gupta, A.; Antes, T.J. Integrated systems for exosome investigation. Methods 2015, 87, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Ferrer-Luna, R.; Rooj, A.K.; Mineo, M.; Ricklefs, F.; Takeda, Y.S.; Nowicki, M.O.; Salinska, E.; Nakano, I.; Lee, H.; et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep. 2017, 8, 1497–1505. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Broadinstitute, Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 1 March 2022).
- Huang, Z.P.; Chen, J.; Seok, H.Y.; Zhang, Z.; Kataoka, M.; Hu, X.; Wang, D.-Z. MicroRNA-22 Regulates Cardiac Hypertrophy and Remodeling in Response to Stress. Circ. Res. 2013, 112, 1234–1243. [Google Scholar] [CrossRef]
- Xiao, C.; Rajewsky, K. MicroRNA Control in the Immune System: Basic Principles. Cell 2009, 136, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Calado, D.P.; Galler, G.; Thai, T.-H.; Patterson, H.C.; Wang, J.; Rajewsky, N.; Bender, T.P. MiR-150 Controls B Cell Differentiation by Targeting the Transcription Factor c-Myb. Cell 2007, 131, 146–159. [Google Scholar] [CrossRef]
- Bao, M.H.; Feng, X.; Zhang, Y.W.; Lou, X.Y.; Cheng, Y.; Zhou, H.H. Let-7 in Cardiovascular Diseases, Heart Development and Cardiovascular Differentiation from Stem Cells. Int. J. Mol. Sci. 2013, 14, 23086–23102. [Google Scholar] [CrossRef]
- Mun, D.; Kim, H.; Kang, J.; Park, H.; Park, H.; Lee, S.; Yun, N.; Joung, B. Expression of miRNAs in circulating exosomes derived from patients with persistent atrial fibrillation. FASEB J. 2019, 33, 5979–5989. [Google Scholar] [CrossRef]
- Aday, S.; Hazan-Halevy, I.; Chamorro-Jorganes, A.; Anwar, M.; Goldsmith, M.; Beazley-Long, N.; Sahoo, S.; Dogra, N.; Sweaad, W.; Catapano, F.; et al. Bioinspired artificial exosomes based on lipid nanoparticles carrying let-7b-5p promote angiogenesis in vitro and in vivo. Mol. Ther. 2021, 29, 2239–2252. [Google Scholar] [CrossRef]
- Chi, N.; Chiou, H.; Chou, S.; Hu, C.; Chen, K.; Chang, C.; Hsieh, Y. Hyperglycemia-related FAS gene and hsa-let-7b-5p as markers of poor outcomes for ischaemic stroke. Eur. J. Neurol. 2020, 27, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Munshi, S.U.; Panda, H.; Holla, P.; Rewari, B.B.; Jameel, S. MicroRNA-150 Is a Potential Biomarker of HIV/AIDS Disease Progression and Therapy. PLoS ONE 2014, 9, e95920. [Google Scholar] [CrossRef]
- Goretti, E.; Rolland-Turner, M.; Léonard, F.; Zhang, L.; Wagner, D.R.; Devaux, Y. MicroRNA-16 affects key functions of human endothelial progenitor cells. J. Leukoc. Biol. 2013, 93, 645–655. [Google Scholar] [CrossRef]
- Rizzacasa, B.; Morini, E.; Mango, R.; Vancheri, C.; Budassi, S.; Massaro, G.; Maletta, S.; Macrini, M.; D’annibale, S.; Romeo, F.; et al. MiR-423 is differentially expressed in patients with stable and unstable coronary artery disease: A pilot study. PLoS ONE 2019, 14, e0216363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, S.; Muhammad, S.; Ren, Q.; Sun, C. miR-103/107 promote ER stress-mediated apoptosis via targeting the Wnt3a/β-catenin/ATF6 pathway in preadipocytes. J. Lipid Res. 2018, 59, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.P.; Wang, X.H.; Zhang, H.X.; Shang, M.-M.; Liu, X.-X.; Sun, H.-M.; Song, Y.-P. MiR-103 regulates the angiogenesis of ischemic stroke rats by targeting vascular endothelial growth factor (VEGF). Iran. J. Basic Med. Sci. 2018, 21, 318–324. [Google Scholar] [CrossRef]
- Natarelli, L.; Geißler, C.; Csaba, G.; Wei, Y.; Zhu, M.; di Francesco, A.; Hartmann, P.; Zimmer, R.; Schober, A. miR-103 promotes endothelial maladaptation by targeting lncWDR59. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Chen, X.; Zhang, H.; Shi, Z. MiR-103a targeting Piezo1 is involved in acute myocardial infarction through regulating endothelium function. Cardiol. J. 2013, 23, 556–562. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Fonseca Ferreira, A.; Wei, J.; Zhang, L.; Macon, C.J.; Degnan, B.; Jayaweera, D.; Hare, J.M.; Kolber, M.A.; Bellio, M.; Khan, A.; et al. HIV Promotes Atherosclerosis via Circulating Extracellular Vesicle MicroRNAs. Int. J. Mol. Sci. 2023, 24, 7567. https://doi.org/10.3390/ijms24087567
Da Fonseca Ferreira A, Wei J, Zhang L, Macon CJ, Degnan B, Jayaweera D, Hare JM, Kolber MA, Bellio M, Khan A, et al. HIV Promotes Atherosclerosis via Circulating Extracellular Vesicle MicroRNAs. International Journal of Molecular Sciences. 2023; 24(8):7567. https://doi.org/10.3390/ijms24087567
Chicago/Turabian StyleDa Fonseca Ferreira, Andrea, Jianqin Wei, Lukun Zhang, Conrad J. Macon, Bernard Degnan, Dushyantha Jayaweera, Joshua M. Hare, Michael A. Kolber, Michael Bellio, Aisha Khan, and et al. 2023. "HIV Promotes Atherosclerosis via Circulating Extracellular Vesicle MicroRNAs" International Journal of Molecular Sciences 24, no. 8: 7567. https://doi.org/10.3390/ijms24087567
APA StyleDa Fonseca Ferreira, A., Wei, J., Zhang, L., Macon, C. J., Degnan, B., Jayaweera, D., Hare, J. M., Kolber, M. A., Bellio, M., Khan, A., Pan, Y., Dykxhoorn, D. M., Wang, L., & Dong, C. (2023). HIV Promotes Atherosclerosis via Circulating Extracellular Vesicle MicroRNAs. International Journal of Molecular Sciences, 24(8), 7567. https://doi.org/10.3390/ijms24087567