
GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier?



Abstract
1. Introduction
2. GSK-3β Inhibition
2.1. Compounds and State of the Art
2.1.1. Natural Compounds and Derivatives
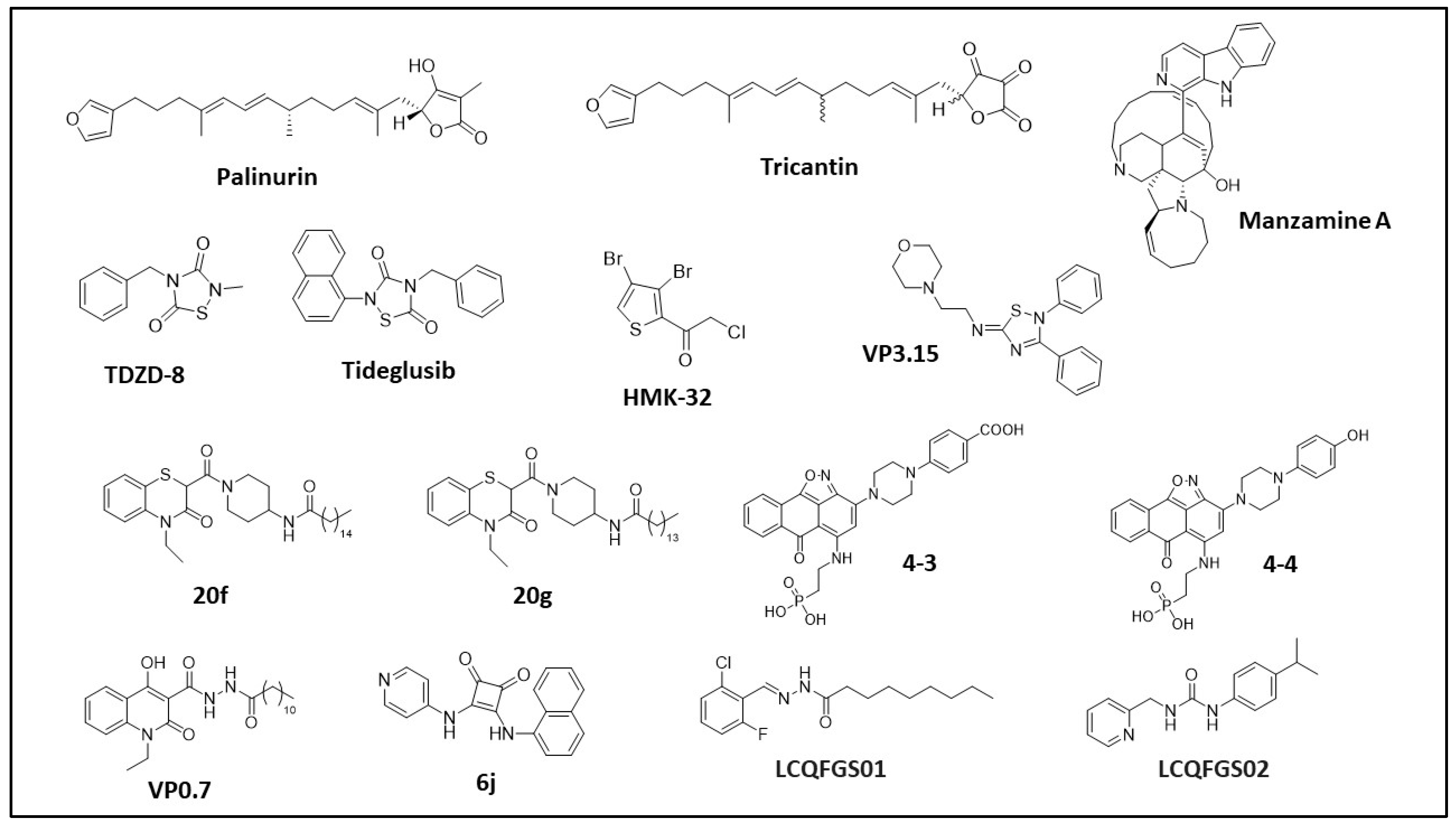
Palinurin and Tricantin
Manzamine Alkaloids
2.1.2. Synthetic Compounds and Derivatives
Thiadiazolidinones and Derivatives (TDZD)
Halomethylketones (HMKs) and Derivatives
5-Imino-1,2,4-Thiadiazoles (ITDZs)
Benzothiazinones (BTOs) and Derivatives
Compounds 4-3 and 4-4
VP0.7 and Derivatives
6j Squaramide
LCQFGS01 and LCQFGS02
3. Discussion and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal Structure of Glycogen Synthase Kinase 3β: Structural Basis for Phosphate-Primed Substrate Specificity and Autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (GSK3): Regulation, Actions, and Diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.; Belmaker, R.H.; Agam, G. The Possible Involvement of Glycogen Synthase Kinase-3 (GSK-3) in Diabetes, Cancer and Central Nervous System Diseases. Curr. Pharm. Des. 2011, 17, 2264–2277. [Google Scholar]
- Muneer, A. Wnt and GSK3 Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications. Clin. Psychopharmacol. Neurosci. 2017, 35, 1761–1774. [Google Scholar] [CrossRef]
- Polter, A.; Beurel, E.; Yang, S.; Garner, R.; Song, L.; Miller, C.A.; Sweatt, J.D.; McMahon, L.; Bartolucci, A.A.; Li, X.; et al. Deficiency in the Inhibitory Serine-Phosphorylation of Glycogen Synthase Kinase-3 Increases Sensitivity to Mood Disturbances. Neuropsychopharmacology 2010, 14, 822–830. [Google Scholar] [CrossRef]
- Beurel, E.; Mines, M.A.; Song, L.; Jope, R.S. Glycogen Synthase Kinase-3 Levels and Phosphorylation Undergo Large Fluctuations in Mouse Brain during Development. Bipolar Disord. 2012, 14, 822–830. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef]
- Li, D.W.; Liu, Z.Q.; Chen, W.; Yao, M.; Li, G.R. Association of glycogen synthase kinase-3β with Parkinson’s disease (review). Mol. Med. Rep. 2014, 9, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; George, J.L.; Wills, J.; Duka, V.; Shah, K.; Lee, Y.C.; Rodriguez, O.; Simkins, T.; Winter, M.; Moechars, D.; et al. GSK-3β Dysregulation Contributes to Parkinson’s-like Pathophysiology with Associated Region-Specific Phosphorylation and Accumulation of Tau and α-Synuclein. Cell Death Differ. 2015, 22, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 beta (GSK-3β) signaling: Implications for Parkinson’s disease. Pharm. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.K.H.; Hung, L.W.; Pang, T.Y.; Mclean, C.A.; Liddell, J.R.; Hilton, J.B.; Li, Q.X.; White, A.R.; Hannan, A.J.; Crouch, P.J. Localized Changes to Glycogen Synthase Kinase-3 and Collapsin Response Mediator Protein-2 in the Huntington’s Disease Affected Brain. Hum. Mol. Genet. 2014, 23, 4051–4063. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-Induced Tau Pathology Drives Hippocampal Neuronal Cell Death in Huntington’s Disease: Involvement of Astrocyte-Neuron Interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Hernández, F.; Miguez, A.; Alberch, J.; Ginés, S.; Pérez-Navarro, E.; Lucas, J.J. Decreased Glycogen Synthase Kinase-3 Levels and Activity Contribute to Huntington’s Disease. Hum. Mol. Genet. 2015, 24, 5040–5052. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in Neurodegenerative Diseases. Int. J. Alzheimer’s Dis. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen Synthase Kinases: Moonlighting Proteins with Theranostic Potential in Cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Fu, Y.; Hu, D.; Qiu, J.; Xie, X.; Ye, F.; Lu, W.G. Overexpression of Glycogen Synthase Kinase-3 in Ovarian Carcinoma Cells with Acquired Paclitaxel Resistance. Int. J. Gynecol. Cancer 2011, 21, 439–444. [Google Scholar] [CrossRef]
- Kawazoe, H.; Bilim, V.N.; Ugolkov, A.V.; Yuuki, K.; Naito, S.; Nagaoka, A.; Kato, T.; Tomita, Y. GSK-3 Inhibition in Vitro and in Vivo Enhances Antitumor Effect of Sorafenib in Renal Cell Carcinoma (RCC). Biochem. Biophys. Res. Commun. 2012, 423, 490–495. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 Suppression Upregulates β-Catenin and c-Myc to Abrogate KRas-Dependent Tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Mccubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as Potential Target for Therapeutic Irvention in Cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef]
- Domoto, T.; Uehara, M.; Bolidong, D.; Minamoto, T. Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells 2020, 9, 1388. [Google Scholar] [CrossRef]
- Masi, M.; Racchi, M.; Travelli, C.; Corsini, E.; Buoso, E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells 2021, 10, 2999. [Google Scholar] [CrossRef]
- Leis, H.; Segrelles, C.; Ruiz, S.; Santos, M.; Paramio, J.M. Expression, Localization, and Activity of Glycogen Synthase Kinase 3β during Mouse Skin Tumorigenesis. Mol. Carcinog. 2002, 35, 180–185. [Google Scholar] [CrossRef]
- Dembowy, J.; Adissu, H.A.; Liu, J.C.; Zacksenhaus, E.; Woodgett, J.R. Effect of Glycogen Synthase Kinase-3 Inactivation on Mouse Mammary Gland Development and Oncogenesis. Oncogene 2015, 34, 3514–3526. [Google Scholar] [CrossRef]
- Shakoori, A.; Mai, W.; Miyashita, K.; Yasumoto, K.; Takahashi, Y.; Ooi, A.; Kawakami, K.; Minamoto, T. Inhibition of GSK-3β Activity Attenuates Proliferation of Human Colon Cancer Cells in Rodents. Cancer Sci. 2007, 98, 1388–1393. [Google Scholar] [CrossRef]
- Garcea, G.; Manson, M.; Neal, C.; Pattenden, C.; Sutton, C.; Dennison, A.; Berry, D. Glycogen Synthase Kinase-3 Beta; A New Target in Pancreatic Cancer? Curr. Cancer Drug Targets 2007, 7, 209–215. [Google Scholar] [CrossRef]
- La Pietra, V.; La Regina, G.; Coluccia, A.; Famiglini, V.; Pelliccia, S.; Plotkin, B.; Eldar-Finkelman, H.; Brancale, A.; Ballatore, C.; Crowe, A.; et al. Design, Synthesis, and Biological Evaluation of 1-Phenylpyrazolo[3,4-e]Pyrrolo[3,4-G]Indolizine-4,6(1 H,5 h)-Diones as New Glycogen Synthase Kinase-3β Inhibitors. J. Med. Chem. 2013, 56, 10066–10078. [Google Scholar] [CrossRef]
- Arfeen, M.; Bhagat, S.; Patel, R.; Prasad, S.; Roy, I.; Chakraborti, A.K.; Bharatam, P.V. Design, Synthesis and Biological Evaluation of 5-Benzylidene-2-Iminothiazolidin-4-Ones as Selective GSK-3β Inhibitors. Eur. J. Med. Chem. 2016, 121, 727–736. [Google Scholar] [CrossRef]
- Ye, Q.; Xu, G.; Lv, D.; Cheng, Z.; Li, J.; Hu, Y. Synthesis and Biological Evaluation of Novel 4-Azaindolyl-Indolyl-Maleimides as Glycogen Synthase Kinase-3β (GSK-3β) Inhibitors. Bioorg. Med. Chem. 2009, 17, 4302–4312. [Google Scholar] [CrossRef]
- Noori, M.S.; Bhatt, P.M.; Courreges, M.C.; Ghazanfari, D.; Cuckler, C.; Orac, C.M.; McMills, M.C.; Schwartz, F.L.; Deosarkar, S.P.; Bergmeier, S.C.; et al. Identification of a Novel Selective and Potent Inhibitor of Glycogen Synthase Kinase-3. Am. J. Physiol.-Cell Physiol. 2019, 317, C1289–C1303. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.X.; Wang, X.L.; Chen, J.Q.; Li, B.; Hur, E.M. Saijilafu Differential Roles of Glycogen Synthase Kinase 3 Subtypes Alpha and Beta in Cortical Development. Front. Mol. Neurosci. 2017, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.; Campillo, N.E. Glycogen Synthase Kinase 3 (GSK-3) Inhibitors: A Patent Update (2016–2019). Expert Opin. Ther. Pat. 2020, 30, 863–872. [Google Scholar] [CrossRef]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the Binding Sites of Glycogen Synthase Kinase 3. Identification and Characterization of Allosteric Modulation Cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef]
- La Sala, G.; Decherchi, S.; De Vivo, M.; Rocchia, W. Allosteric Communication Networks in Proteins Revealed through Pocket Crosstalk Analysis. ACS Cent. Sci. 2017, 3, 949–960. [Google Scholar] [CrossRef]
- Decherchi, S.; Rocchia, W. A General and Robust Ray-Casting-Based Algorithm for Triangulating Surfaces at the Nanoscale. PLoS ONE 2013, 8, e59744. [Google Scholar] [CrossRef]
- Decherchi, S.; Spitaleri, A.; Stone, J.; Rocchia, W. NanoShaper–VMD Interface: Computing and Visualizing Surfaces, Pockets and Channels in Molecular Systems. Bioinformatics 2019, 35, 1241–1243. [Google Scholar] [CrossRef]
- Gagliardi, L.; Rocchia, W. SiteFerret: Beyond Simple Pocket Identification in Proteins. arXiv 2022, arXiv:2212.11888. [Google Scholar]
- Alonso Gordillo, D.; Dorronsoro Diaz, I.; Martinez Gil, A.; Panizo del Pliego, G.; Fuertes Huerta, A.; Perez Puerto, M.J.; Martin Aparicio, E.; Perez Navarro, D.; Medina Padilla, M. GSK-3 Inhibitors Isolated from Marine Organisms. U.S. Patent WO2005054221A1, 16 June 2005. [Google Scholar]
- Alonso, D.; Dorronsoro, I.; Panizo, G.; Martin-Aparicio, E.; Jose Perez-Puerto, M.; Medina, M.; Fernandez-Chimeno, R.I.; Perez-Baz, J.; Martinez, A. P4–335: Glycogen Synthase Kinase–3β Inhibitors from Marine Origin. Alzheimer’s Dement. 2006, 2, 615. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Pérez, M.; Pérez, D.I.; Martínez, A.; Castro, A.; Gómez, G.; Fall, Y. The First Enantioselective Synthesis of Palinurin. Chem. Commun. 2009, 22, 3252–3254. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Caron, G.; Pintos, I.G.; Gerbaldo, M.; Pérez, M.; Pérez, D.I.; Gándara, Z.; Martínez, A.; Gómez, G.; Fall, Y. An Application of Two MIFs-Based Tools (Volsurf+ and Pentacle) to Binary QSAR: The Case of a Palinurin-Related Data Set of Non-ATP Competitive Glycogen Synthase Kinase 3β (GSK-3β) Inhibitors. Eur. J. Med. Chem. 2011, 46, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a New Binding Mode to GSK-3: Allosteric Regulation by the Marine Compound Palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Gelpí, J.L.; Kalko, S.G.; Barril, X.; Cirera, J.; de La Cruz, X.; Luque, F.J.; Orozco, M. Classical Molecular Interaction Potentials: Improved Setup Procedure in Molecular Dynamics Simulations of Proteins. Proteins Struct. Funct. Genet. 2001, 45, 428–437. [Google Scholar] [CrossRef]
- Sakai, R.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Manzamine A, a Novel Antitumor Alkaloid from a Sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Yousaf, M.; Hammond, N.L.; Peng, J.; Wahyuono, S.; McIntosh, K.A.; Charman, W.N.; Mayer, A.M.S.; Hamann, M.T. New Manzamine Alkaloids from an Indo-Pacific Sponge. Pharmacokinetics, Oral Availability, and the Significant Activity of Several Manzamines against HIV-I, AIDS Opportunistic Infections, and Inflammatory Diseases. J. Med. Chem. 2004, 47, 3512–3517. [Google Scholar] [CrossRef]
- Rao, K.V.; Donia, M.S.; Peng, J.; Garcia-Palomero, E.; Alonso, D.; Martinez, A.; Medina, M.; Franzblau, S.G.; Tekwani, B.L.; Khan, S.I.; et al. Manzamine B and E and Ircinal A Related Alkaloids from an Indonesian Acanthostrongylophora Sponge and Their Activity against Infectious, Tropical Parasitic, and Alzheimer’s Diseases. J. Nat. Prod. 2006, 69, 1034–1040. [Google Scholar] [CrossRef]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; del Monte-Millán, M.; Medina, M.; et al. Glycogen Synthase Kinase-3 (GSK-3) Inhibitory Activity and Structure-Activity Relationship (SAR) Studies of the Manzamine Alkaloids. Potential for Alzheimer’s Disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef]
- Lin, L.-C.; Kuo, T.-T.; Chang, H.-Y.; Liu, W.-S.; Hsia, S.-M.; Huang, T.-C. Manzamine A Exerts Anticancer Activity against Human Colorectal Cancer Cells. Mar. Drugs 2018, 16, 252. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.A.; Johnson, J.D.; Linley, P.A.; Gunasekera, S.E.; Wright, A.E. A Novel Activity from an Old Compound: Manzamine A Reduces the Metastatic Potential of AsPC-1 Pancreatic Cancer Cells and Sensitizes Them to TRAIL-Induced Apoptosis. Investig. New Drugs 2011, 29, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Mamaghani, S.; Simpson, C.D.; Cao, P.M.; Cheung, M.; Chow, S.; Bandarchi, B.; Schimmer, A.D.; Hedley, D.W. Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to TRAIL-Induced Apoptosis. PLoS ONE 2012, 7, e41102. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kudrimoti, S.; Prasanna, S.; Odde, S.; Doerksen, R.J.; Pennaka, H.K.; Choo, Y.M.; Rao, K.V.; Tekwani, B.L.; Madgula, V.; et al. Structure—Activity Relationship and Mechanism of Action Studies of Manzamine Analogues for the Control of Neuroinflammation and Cerebral Infections. J. Med. Chem. 2010, 53, 61–76. [Google Scholar] [CrossRef]
- Osolodkin, D.I.; Shulga, D.A.; Palyulin, V.A.; Zefirov, N.S. Interaction of Manzamine A with Glycogen Synthase Kinase 3β: A Molecular Dynamics Study. Russ. Chem. Bull. 2010, 59, 1983–1993. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Martínez, A.; Castro, A.; Cardelús, I.; Llenas, J.; Palacios, J.M. Arylimino-1,2,4-Thiadiazolidinones: A New Family of Potassium Channel Openers. Bioorg. Med. Chem. 1997, 5, 1275–1283. [Google Scholar] [CrossRef]
- Martinez, A.; Fernandez, E.; Castro, A.; Conde, S.; Rodriguez-Franco, I.; Baos, J.E.; Badia, A. N-Benzylpiperidine Derivatives of 1,2,4-Thiadiazolidinone as New Acetylcholinesterase Inhibitors. Eur. J. Med. Chem. 2000, 35, 913–922. [Google Scholar] [CrossRef]
- Lipina, T.V.; Kaidanovich-Beilin, O.; Patel, S.; Wang, M.; Clapcote, S.J.; Liu, F.; Woodgett, J.R.; Roder, J.C. Genetic and Pharmacological Evidence for Schizophrenia-Related Disc1 Interaction with GSK-3. Synapse 2011, 65, 234–248. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; Kockeritz, L.; Woodgett, J.R.; Gainetdinov, R.R.; Caron, M.G. Lithium Antagonizes Dopamine-Dependent Behaviors Mediated by an AKT/Glycogen Synthase Kinase 3 Signaling Cascade. Proc. Natl. Acad. Sci. USA 2004, 101, 5099–5104. [Google Scholar] [CrossRef]
- Huang, S.; Wang, H.; Turlova, E.; Abussaud, A.; Ji, X.; Britto, L.R.; Miller, S.P.; Martinez, A.; Sun, H.S.; Feng, Z.P. GSK-3β Inhibitor TDZD-8 Reduces Neonatal Hypoxic-Ischemic Brain Injury in Mice. CNS Neurosci Ther. 2017, 23, 405–415. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Altieri, D.C. Activation of P53-Dependent Apoptosis by Acute Ablation of Glycogen Synthase Kinase-3β in Colorectal Cancer Cells. Clin. Cancer Res. 2005, 11, 4580–4588. [Google Scholar] [CrossRef]
- Guzman, M.L.; Li, X.; Corbett, C.A.; Rossi, R.M.; Bushnell, T.; Liesveld, J.L.; Hé, J.; Young, F.; Jordan, C.T. Rapid and Selective Death of Leukemia Stem and Progenitor Cells Induced by the Compound 4-Benzyl, 2-Methyl, 1,2,4-Thiadiazolidine, 3,5 Dione (TDZD-8). Blood 2007, 110, 4436–4444. [Google Scholar] [CrossRef]
- Aguilar-Morante, D.; Morales-Garcia, J.A.; Sanz-Sancristobal, M.; Garcia-Cabezas, M.A.; Santos, A.; Perez-Castillo, A. Inhibition of Glioblastoma Growth by the Thiadiazolidinone Compound TDZD-8. PLoS ONE 2010, 5, e13879. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of Glycogen Synthase Kinase 3 Activity Reduces Tumor Growth of Prostate Cancer in Vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, X.; Cheng, X.; Ye, F.; Xie, X.; Lu, W. Clinicopathological and Biological Significance of Aberrant Activation of Glycogen Synthase Kinase-3 in Ovarian Cancer. Onco Targets Ther. 2014, 7, 1159–1168. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen Synthase Kinase 3β (GSK3β) Mediates 6- Hydroxydopamine-induced Neuronal Death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef]
- Xie, C.L.; Lin, J.Y.; Wang, M.H.; Zhang, Y.; Zhang, S.F.; Wang, X.J.; Liu, Z.G. Inhibition of Glycogen Synthase Kinase-3β (GSK-3β) as Potent Therapeutic Strategy to Ameliorates L-Dopa-Induced Dyskinesia in 6-OHDA Parkinsonian Rats. Sci. Rep. 2016, 6, 23527. [Google Scholar] [CrossRef]
- Koehler, D.; Shah, Z.A.; Williams, F.E. The GSK3β Inhibitor, TDZD-8, Rescues Cognition in a Zebrafish Model of Okadaic Acid-Induced Alzheimer’s Disease. Neurochem. Int. 2019, 122, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Ju, H.; Zhou, Y.; Yang, Y.; Shi, Y.; Fang, H. Inhibition of Glycogen Synthase Kinase 3β Improves Cognitive Function in Aged Mice by Upregulating Claudin Presences in Cerebral Endothelial Cells. Acta Biochim. Biophys. Sin. 2021, 52, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. A-Synuclein Contributes to GSK-3β-catalyzed Tau Phosphorylation in Parkinson’s Disease Models. FASEB J. 2009, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cochemé, H.M.; et al. Direct Keap1-Nrf2 Disruption as a Potential Therapeutic Target for Alzheimer’s Disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-Mediated Activation of GSK3β Promotes Tau Phosphorylation and Impairs Memory in Type 2 Diabetes. Neurobiol. Aging 2017, 57, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, J.; Zeng, J.; Hu, B.; Fang, X.; Li, L. Inhibition of GSK-3β Alleviates Collagen II-Induced Rheumatoid Arthritis in Rats. Med. Sci. Monit. 2016, 22, 1047–1052. [Google Scholar] [CrossRef]
- Mateus, V.; Rocha, J.; Alves, P.; Mota-Filipe, H.; Sepodes, B.; Pinto, R. Thiadiazolidinone-8 Ameliorates Inflammation Associated with Experimental Colitis in Mice. Pharmacology 2017, 101, 35–42. [Google Scholar] [CrossRef]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A Novel GSK-3β Inhibitor Reduces Alzheimer’s Pathology and Rescues Neuronal Loss In Vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Domínguez, J.M.; Fuertes, A.; Orozco, L.; del Monte-Millán, M.; Delgado, E.; Medina, M. Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3β by Tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J. Alzheimer’s Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Huppertz, H.J.; Wagenpfeil, S.; Andrés, M.V.; Belloch, V.; León, T.; del Ser, T. Tideglusib Reduces Progression of Brain Atrophy in Progressive Supranuclear Palsy in a Randomized Trial. Mov. Disord. 2014, 29, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Litvan, I.; Höglinger, G.U.; Burn, D.J.; Lees, A.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; del Ser, T.; Gómez, J.C.; et al. A Phase 2 Trial of the GSK-3 Inhibitor Tideglusib in Progressive Supranuclear Palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Chauhan, N.; Gajjar, A.; Basha, S.H. Pharmacophore Feature-Based Virtual Screening for Finding Potent GSK-3 Inhibitors Using Molecular Docking and Dynamics Simulations. Bioinformation 2016, 12, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Conde, S.; Pérez, D.I.; Martínez, A.; Perez, C.; Moreno, F.J. Thienyl and Phenyl α-Halomethyl Ketones: New Inhibitors of Glycogen Synthase Kinase (GSK-3β) from a Library of Compound Searching. J. Med. Chem. 2003, 46, 4631–4633. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.I.; Conde, S.; Pérez, C.; Gil, C.; Simon, D.; Wandosell, F.; Moreno, F.J.; Gelpí, J.L.; Luque, F.J.; Martínez, A. Thienylhalomethylketones: Irreversible Glycogen Synthase Kinase 3 Inhibitors as Useful Pharmacological Tools. Bioorg. Med. Chem. 2009, 17, 6914–6925. [Google Scholar] [CrossRef]
- Perez, D.I.; Palomo, V.; Pérez, C.; Gil, C.; Dans, P.D.; Luque, F.J.; Conde, S.; Martínez, A. Switching Reversibility to Irreversibility in Glycogen Synthase Kinase 3 Inhibitors: Clues for Specific Design of New Compounds. J. Med. Chem. 2011, 54, 4042–4056. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Perez, C.; Morales-Garcia, J.A.; Soteras, I.; Alonso-Gil, S.; Encinas, A.; Castro, A.; Campillo, N.E.; Perez-Castillo, A.; et al. 5-Imino-1,2,4-Thiadiazoles: First Small Molecules As Substrate Competitive Inhibitors of Glycogen Synthase Kinase 3. J. Med. Chem. 2012, 55, 1645–1661. [Google Scholar] [CrossRef]
- Medina-Rodríguez, E.M.; Bribián, A.; Boyd, A.; Palomo, V.; Pastor, J.; Lagares, A.; Gil, C.; Martínez, A.; Williams, A.; de Castro, F. Promoting in Vivo Remyelination with Small Molecules: A Neuroreparative Pharmacological Treatment for Multiple Sclerosis. Sci. Rep. 2017, 7, srep43545. [Google Scholar] [CrossRef]
- Sánchez-Cruz, A.; Villarejo-Zori, B.; Marchena, M.; Zaldivar-Díez, J.; Palomo, V.; Gil, C.; Lizasoain, I.; de La Villa, P.; Martínez, A.; de La Rosa, E.J.; et al. Modulation of GSK-3 Provides Cellular and Functional Neuroprotection in the Rd10 Mouse Model of Retinitis Pigmentosa. Mol. Neurodegener. 2018, 13, 19. [Google Scholar] [CrossRef]
- Benítez-Fernández, R.; Melero-Jerez, C.; Gil, C.; de la Rosa, E.J.; Martínez, A.; de Castro, F. Dynamics of Central Remyelination and Treatment Evolution in a Model of Multiple Sclerosis with Optic Coherence Tomography. Int. J. Mol. Sci. 2021, 22, 2440. [Google Scholar] [CrossRef]
- Sánchez-Cruz, A.; Martínez, A.; de la Rosa, E.J.; Hernández-Sánchez, C. GSK-3 Inhibitors: From the Brain to the Retina and Back Again. In Adv. Exp. Med. Biol. 2019, 1185, 437–441. [Google Scholar]
- Zhang, P.; Hu, H.R.; Huang, Z.H.; Lei, J.Y.; Chu, Y.; Ye, D.Y. Identification of Novel Scaffold of Benzothiazepinones as Non-ATP Competitive Glycogen Synthase Kinase-3β Inhibitors through Virtual Screening. Bioorg. Med. Chem. Lett 2012, 22, 7232–7236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, H.R.; Bian, S.H.; Huang, Z.H.; Chu, Y.; Ye, D.Y. Design, Synthesis and Biological Evaluation of Benzothiazepinones (BTZs) as Novel Non-ATP Competitive Inhibitors of Glycogen Synthase Kinase-3β (GSK-3β). Eur. J. Med. Chem. 2013, 61, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, S.; Gao, Y.; Lu, W.; Huang, K.; Ye, D.; Li, X.; Chu, Y. Novel Benzothiazinones (BTOs) as Allosteric Modulator or Substrate Competitive Inhibitor of Glycogen Synthase Kinase 3β (GSK-3β) with Cellular Activity of Promoting Glucose Uptake. Bioorg. Med. Chem. Lett 2014, 24, 5639–5643. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ye, D.Y.; Zhou, W.C.; Chu, Y. The Discovery of Novel Benzothiazinones as Highly Selective Non-ATP Competitive Glycogen Synthase Kinase 3β Inhibitors for the Treatment of Ovarian Cancer. Eur. J. Med. Chem. 2017, 135, 370–381. [Google Scholar] [CrossRef]
- Cao, Q.; Lu, X.; Feng, Y.J. Glycogen Synthase Kinase-3β Positively Regulates the Proliferation of Human Ovarian Cancer Cells. Cell Res 2006, 16, 671–677. [Google Scholar] [CrossRef]
- Rico, A.; Guembelzu, G.; Palomo, V.; Martínez, A.; Aiastui, A.; Casas-fraile, L.; Valls, A.; de Munain, A.L.; Sáenz, A. Allosteric Modulation of GSK-3β as a New Therapeutic Approach in Limb Girdle Muscular Dystrophy R1 Calpain 3-related. Int. J. Mol. Sci. 2021, 22, 7367. [Google Scholar] [CrossRef]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.-I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 Cells and Experimental Autoimmune Encephalomyelitis by Glycogen Synthase Kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Carullo, G.; Bottoni, L.; Pasquini, S.; Papa, A.; Contri, C.; Brogi, S.; Calderone, V.; Orlandini, M.; Gemma, S.; Varani, K.; et al. Front Cover: Synthesis of Unsymmetrical Squaramides as Allosteric GSK-3β Inhibitors Promoting Β-Catenin-Mediated Transcription of TCF/LEF in Retinal Pigment Epithelial Cells. ChemMedChem 2022, 17, e202200663. [Google Scholar] [CrossRef]
- Silva, G.M.; Borges, R.S.; Santos, K.L.B.; Federico, L.B.; Francischini, I.A.G.; Gomes, S.Q.; Barcelos, M.P.; Silva, R.C.; Santos, C.B.R.; Silva, C.H.T.P. Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8252. [Google Scholar] [CrossRef]
- Rippin, I.; Khazanov, N.; Shirley Ben Joseph Kudinov, T.; Berent, E.; Arciniegas Ruiz, S.M.; Marciano, D.; Levy, L.; Gruzman, A.; Senderowitz, H.; Eldar-Finkelman, H. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. Int. J. Mol. Sci. 2020, 21, 8709. [Google Scholar] [CrossRef]
- Licht-Murava, A.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. Elucidating Substrate and Inhibitor Binding Sites on the Surface of GSK-3β and the Refinement of a Competitive Inhibitor. J. Mol. Biol. 2011, 408, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A Unique Type of GSK-3 Inhibitor Brings New Opportunities to the Clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef]
- Lodish, M.B. Clinical review: Kinase inhibitors: Adverse effects related to the endocrine system. J. Clin. Endocrinol. Metab. 2013, 98, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Laufkötter, O.; Hu, H.; Miljković, F.; Bajorath, J. Structure- and Similarity-Based Survey of Allosteric Kinase Inhibitors, Activators, and Closely Related Compounds. J. Med. Chem. 2021, 65, 922–934. [Google Scholar] [CrossRef]
- Balboni, B.; Tripathi, S.K.; Veronesi, M.; Russo, D.; Penna, I.; Giabbai, B.; Bandiera, T.; Storici, P.; Girotto, S.; Cavalli, A. Identification of Novel GSK-3β Hits Using Competitive Biophysical Assays. Int. J. Mol. Sci. 2022, 23, 3856. [Google Scholar] [CrossRef]
- di Fruscia, P.; Edfeldt, F.; Shamovsky, I.; Collie, G.W.; Aagaard, A.; Barlind, L.; Börjesson, U.; Hansson, E.L.; Lewis, R.J.; Nilsson, M.K.; et al. Fragment-Based Discovery of Novel Allosteric MEK1 Binders. ACS Med. Chem. Lett. 2021, 12, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.; Gajjar, A. Classifying Druggability on Potential Binding Sites of Glycogen Synthase Kinase-3β: An in-Silico Assessment. Acta Pharm. Sci. 2017, 55, 43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
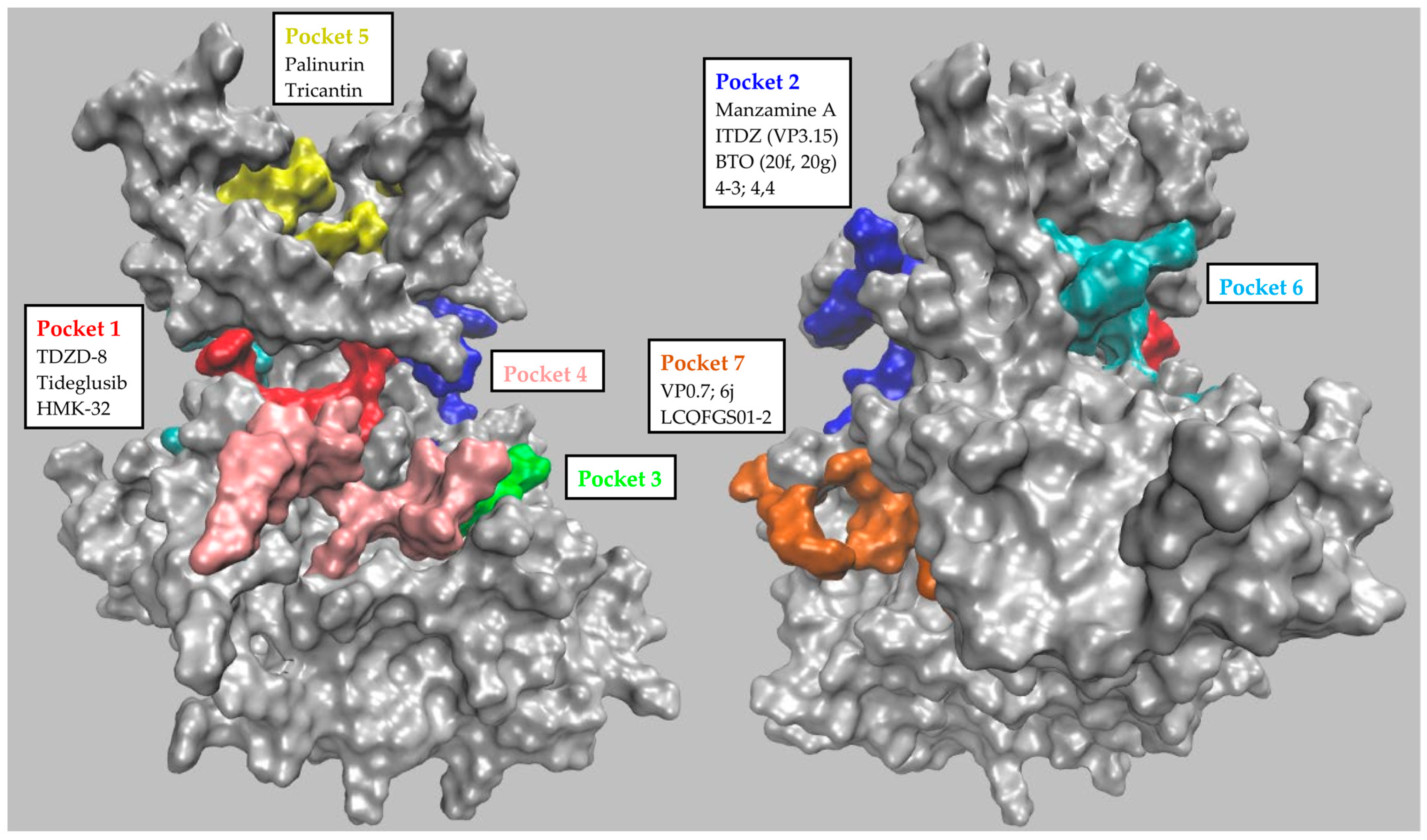
| GSK-3β Pocket | Pocket Key Residues | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Numbering of Palomo et al. | P1 | 85 | 97 | 113 | 134 | 135 | 138 | 186 | 188 | 199 | 200 |
| Residue’s identity | Lys | Glu | Asp | Tyr | Val | Thr | Asn | Leu | Cys | Asp | |
| 1J1C_B numbering | 563 | 575 | 591 | 612 | 613 | 616 | 664 | 666 | 677 | 678 | |
| Numbering of Palomo et al. | P2 | 67 | 89 | 94 | 93 | 95 | 96 | 97 | 180 | 205 | |
| Residue’s identity | Phe | Gln | Lys | Phe | Asn | Arg | Glu | Arg | Lys | ||
| 1J1C_B numbering | 545 | 567 | 572 | 571 | 573 | 574 | 575 | 658 | 683 | ||
| Numbering of Palomo et al. | P3 | 215 | 220 | 223 | 229 | ||||||
| Residue’s identity | Ser | Arg | Arg | Phe | |||||||
| 1J1C_B numbering | 693 | 698 | 701 | 707 | |||||||
| Numbering of Palomo et al. | P4 | 140 | 144 | 148 | 185 | 219 | 220 | 221 | 222 | 249 | |
| Residue’s identity | Tyr | Arg | Arg | Gln | Ser | Arg | Tyr | Tyr | Glu | ||
| 1J1C_B numbering | 618 | 622 | 626 | 663 | 697 | 698 | 699 | 700 | 727 | ||
| Numbering of Palomo et al. | P5 | 26 | 38 | 56 | 71 | 86 | 119 | ||||
| Residue’s identity | Met | Thr | Tyr | Tyr | Lys | Ser | |||||
| 1J1C_B numbering | 504 | 516 | 534 | 549 | 564 | 597 | |||||
| Numbering of Palomo et al. | P6 | 80 | 111 | 113 | 133 | 135 | 190 | 197 | |||
| Residue’s identity | Glu | Arg | Arg | Asp | Val | Asp | Lys | ||||
| 1J1C_B numbering | 558 | 589 | 591 | 611 | 613 | 668 | 675 | ||||
| Numbering of Palomo et al. | P7 | 173 | 178 | 207 | 209 | 211 | 234 | 235 | 236 | 330 | 369 |
| Residue’s identity | His | Cys | Leu | Arg | Glu | Tyr | Thr | Ser | Thr | Ser | |
| 1J1C_B numbering | 651 | 656 | 685 | 687 | 689 | 712 | 713 | 714 | 808 | 847 | |
| Inhibitor | Potency (Kd/IC50) | In Vitro Assays | In Cellulo/Ex Vivo Assays | In Vivo/Clinical Trial | Pocket (Docking) | Preclinical Models | Ref. |
|---|---|---|---|---|---|---|---|
| Palinurin Tricantin | 4.5 μM 7.5 μM | 33P-γ-ATP-based GSK-3β inhibition assay | Inhibition of tau phosphorylation; cell viability | - | Pocket 5 | AD | [42,43,44,45,46,47] |
| Manzamine A | 10.0 μM | 33P-γ-ATP-based GSK-3β inhibition assay | Inhibition of tau phosphorylation; cell viability, proliferation and invasion; apoptosis induction; anti-neuroinflammatory activity | Pharmacokinetic studies; tumor growth inhibition | Pocket 2 (substrate pocket) | AD, neuroinflammation, PaC, PC and CRC | [48,49,50,51,52,53,54,55,56] |
| TDZD-8 | 2.0 μM | 32P-γ-ATP-based GSK-3β inhibition assay | p53 induction; apoptosis analysis; cell viability, toxicity and proliferation; oxidative stress evaluation; inhibition of tau phosphorylation; | Locomotor activity; behavioral studies; attention deficits; information processing; ischemic-induced brain injury; apoptosis suppression; neuronal protection; tumor growth inhibition; effect on joint injury; inflammation effect | Pocket 1 (ATP pocket) | AD, CRC, glioblastoma, PC, PD, OC, IBI, leukemia, schizophrenia, type II diabetes, RA, colitis | [57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77] |
| Tideglusib | - | FRET-based GSK-3β inhibition assay; GSK-3β reversibility inhibition studies; 35S-tideglusib GSK-3β interaction assay | - | ARGO Phase II clinical trial; TAUROS Phase II clinical trial; escalating dose pilot trial | Pocket 1 (ATP pocket) | - | [78,79,80,81,82,83] |
| HMK-32 | 1.5 μM | 32P-γ-ATP-GSK-3β inhibition assay; 32P-γ-ATP-based GSK-3β reversibility inhibition studies | Inhibition of tau phosphorylation; neuritogenesis evaluation | - | Pocket 1 (ATP pocket) | AD | [84,85,86] |
| ITDZ (VP3.15) | 0.9 μM | Luminescence-based GSK-3β inhibition assay; GSK-3β enzymatic inhibition; luminescence-based GSK-3β reversibility studies | Neuroprotection and neurogenesis assessment; BBB penetration; cell survival, proliferation and differentiation; ex vivo remyelination | Remyelination; pharmacokinetic studies; inflammatory and degenerative signs; visual function; EAE clinical signs examination | Pocket 2 (Substrate Pocket) | LGMD, AE, MS, RP | [87,88,89,90,91,92,93] |
| BTO (20f, 20g) | 11.7 μM 22.4 μM | Luminescence-based GSK-3β inhibition assay | Cell viability; cell cycle analysis; apoptosis induction | Tumor growth inhibition | Pocket 2 (Substrate Pocket) | OC | [94,95,96,97] |
| 4-3 4-4 | 1.0–4.0 μM | ELISA-based GSK-3β inhibition assay | Wnt/β-catenin pathway evaluation | - | Pocket 2 (Substrate Pocket) | AD | [98,99,100] |
| VP0.7 | 3.0 μM | Luminescence-based GSK-3β inhibition assay | Wnt/β-catenin pathway evaluation | EAE clinical signs examination | Pocket 7 | AE | [37,98,99,100] |
| 6j | 3.5 μM | FRET-based GSK-3β inhibition assay | Cell viability; β-catenin-mediated TCF/LEF transcriptional activity | - | Pocket 7 | Retinal epithelium | [101] |
| LCQFGS01 LCQFGS02 | - | - | - | - | Pocket 7 | In silico evaluation only | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balboni, B.; Masi, M.; Rocchia, W.; Girotto, S.; Cavalli, A. GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier? Int. J. Mol. Sci. 2023, 24, 7541. https://doi.org/10.3390/ijms24087541
Balboni B, Masi M, Rocchia W, Girotto S, Cavalli A. GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier? International Journal of Molecular Sciences. 2023; 24(8):7541. https://doi.org/10.3390/ijms24087541
Chicago/Turabian StyleBalboni, Beatrice, Mirco Masi, Walter Rocchia, Stefania Girotto, and Andrea Cavalli. 2023. "GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier?" International Journal of Molecular Sciences 24, no. 8: 7541. https://doi.org/10.3390/ijms24087541
APA StyleBalboni, B., Masi, M., Rocchia, W., Girotto, S., & Cavalli, A. (2023). GSK-3β Allosteric Inhibition: A Dead End or a New Pharmacological Frontier? International Journal of Molecular Sciences, 24(8), 7541. https://doi.org/10.3390/ijms24087541