


Finding New Molecular Targets of Two Copper(II)-Hydrazone Complexes on Triple-Negative Breast Cancer Cells Using Mass-Spectrometry-Based Quantitative Proteomics

 and
and 
Abstract
1. Introduction
2. Results and Discussion
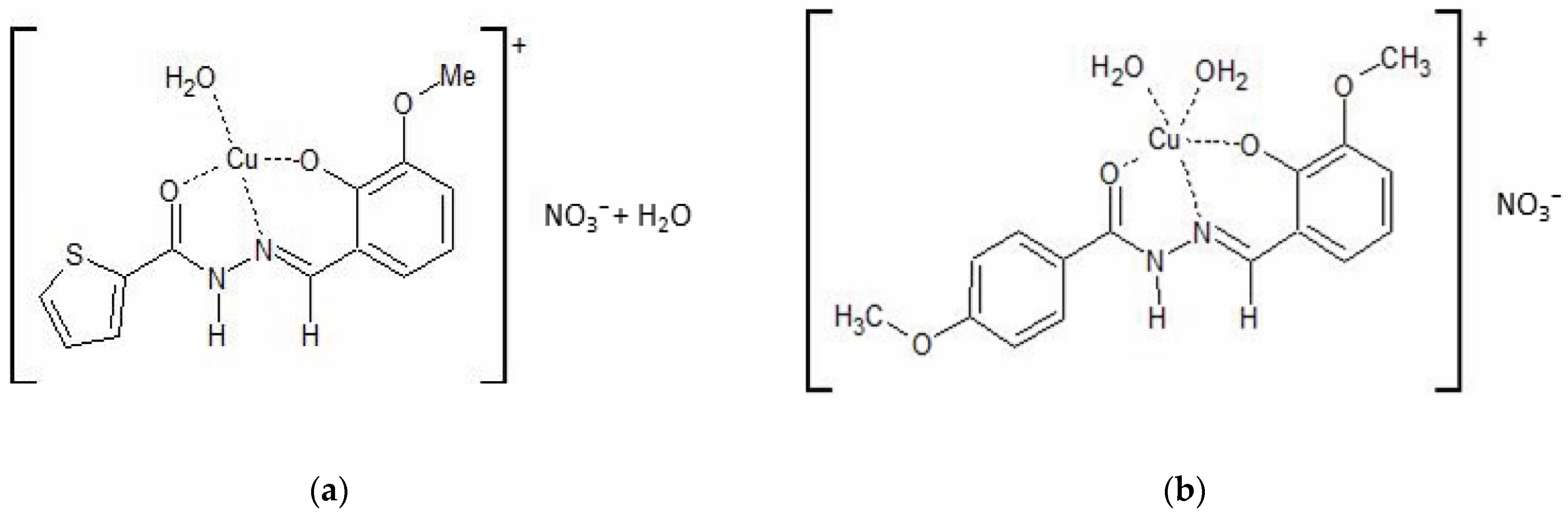
2.1. Synthesis and Characterization of the Copper Complexes
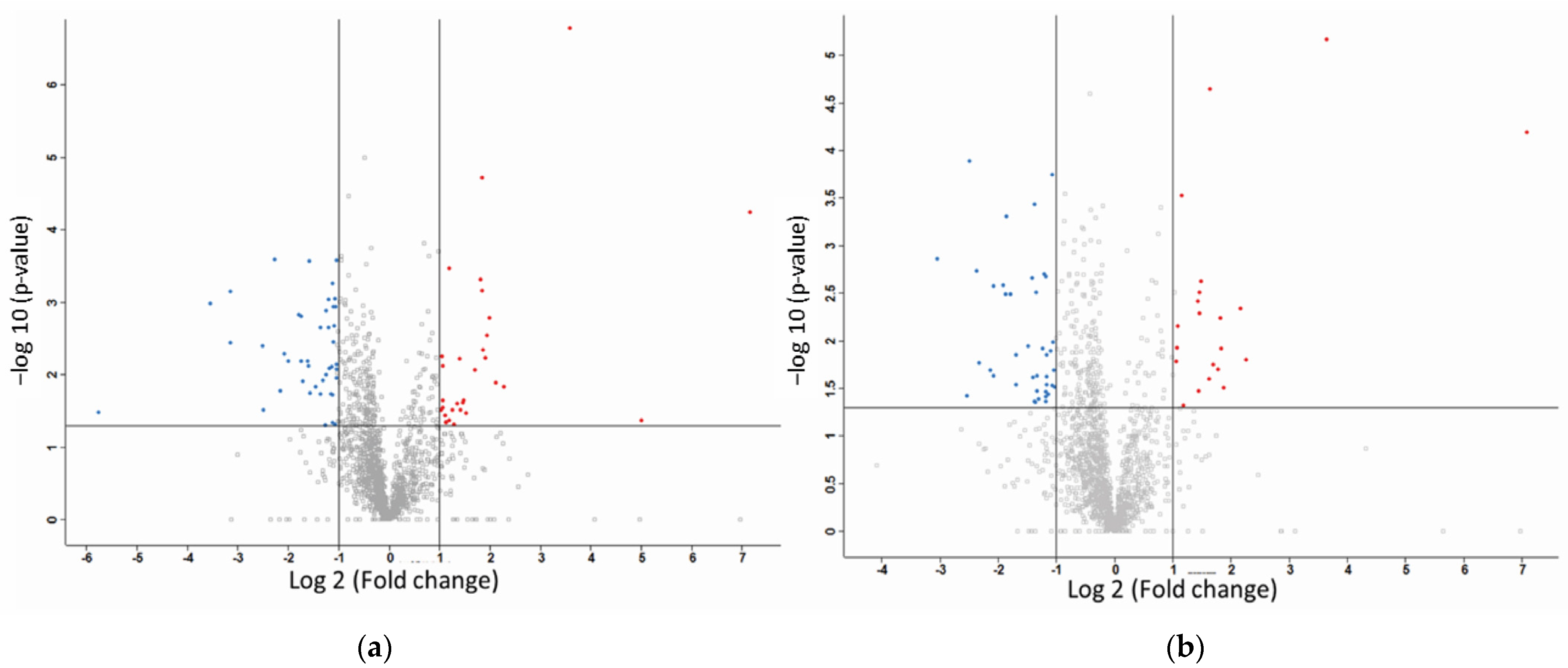
2.2. Label-Free Mass Spectroscopy Quantification of Proteins Isolated from MDA-MB-231 Cells following Treatment with CuHL1 and CuHL2
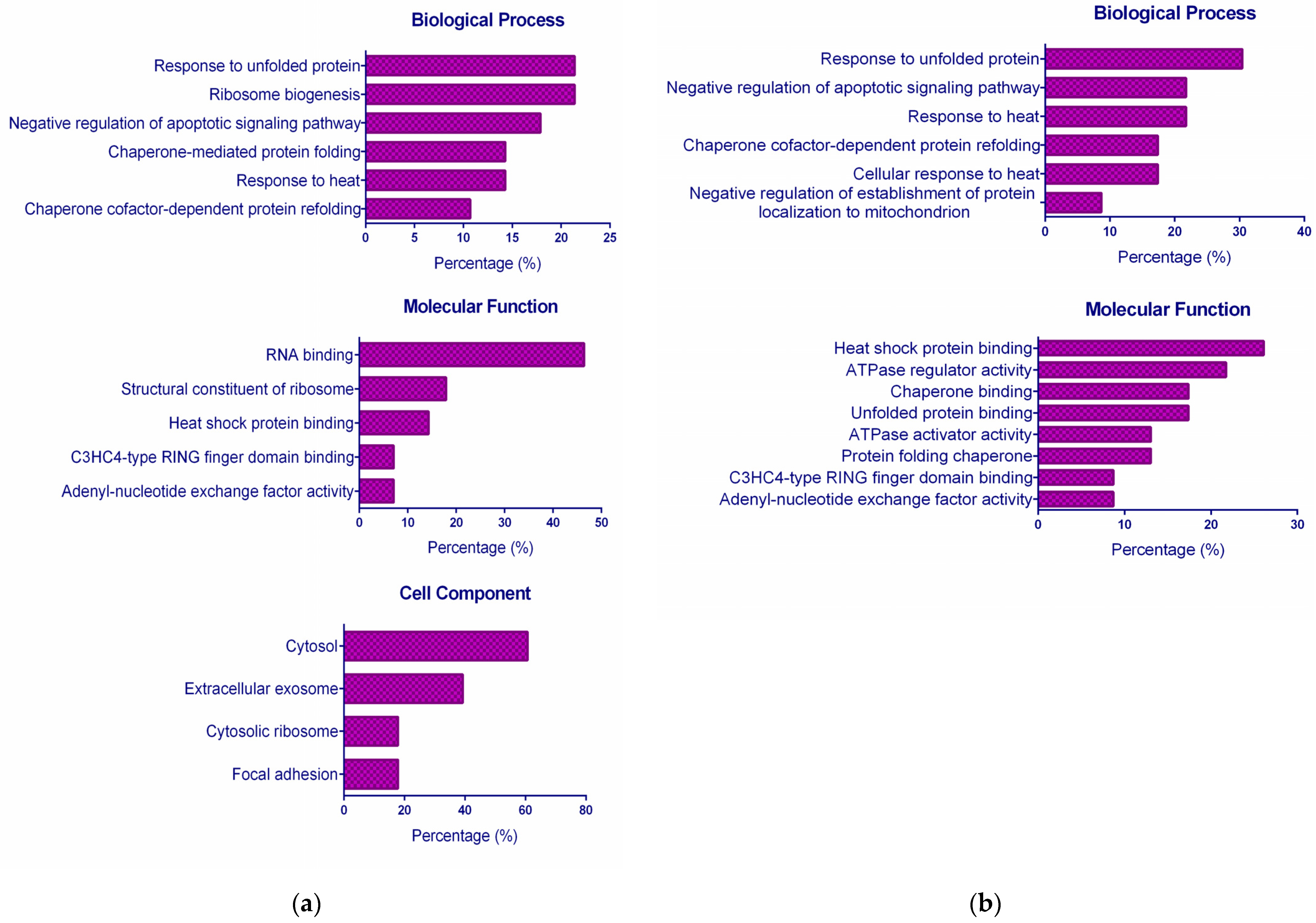
2.3. Functional GO Enrichment Analysis
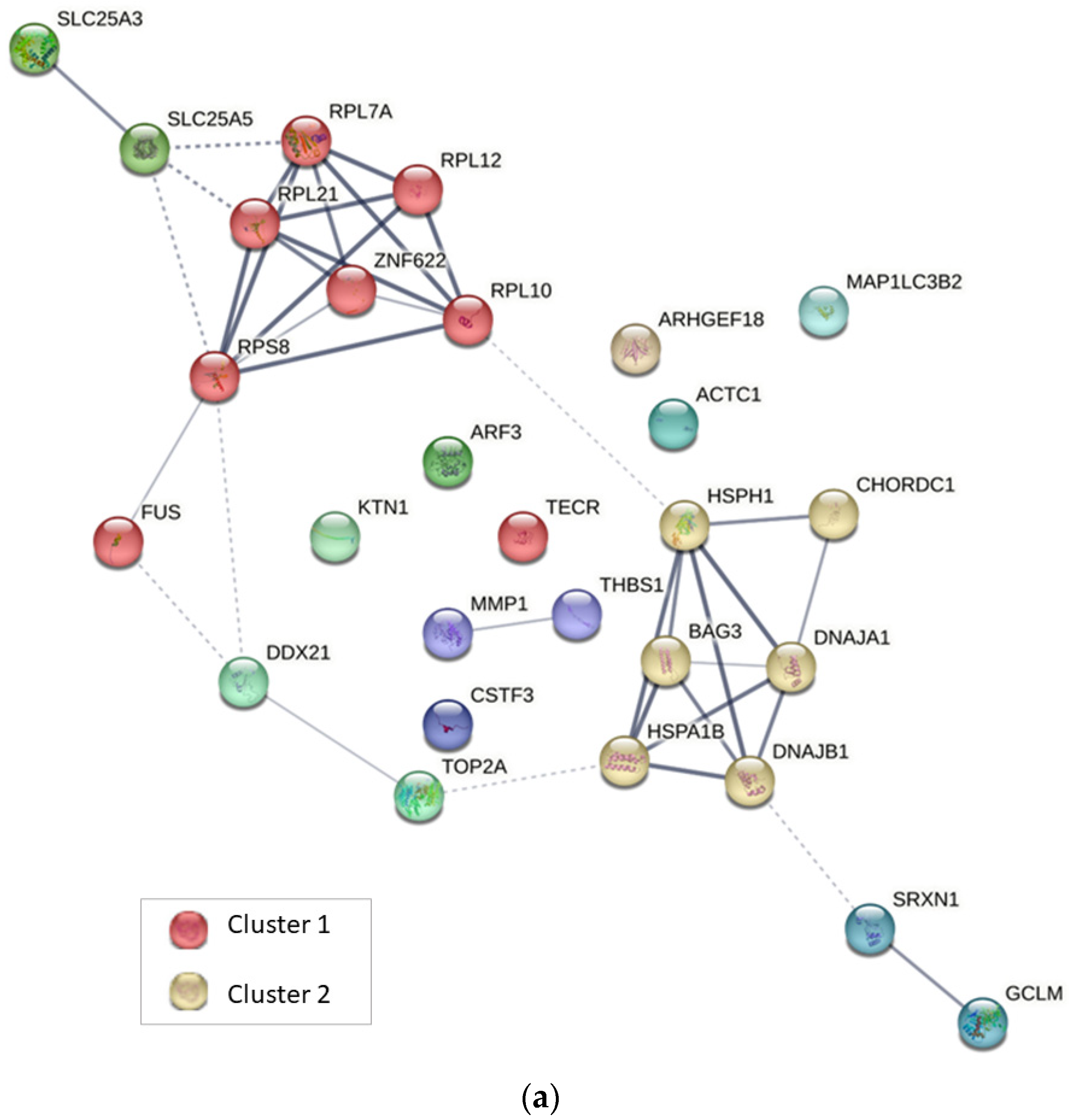
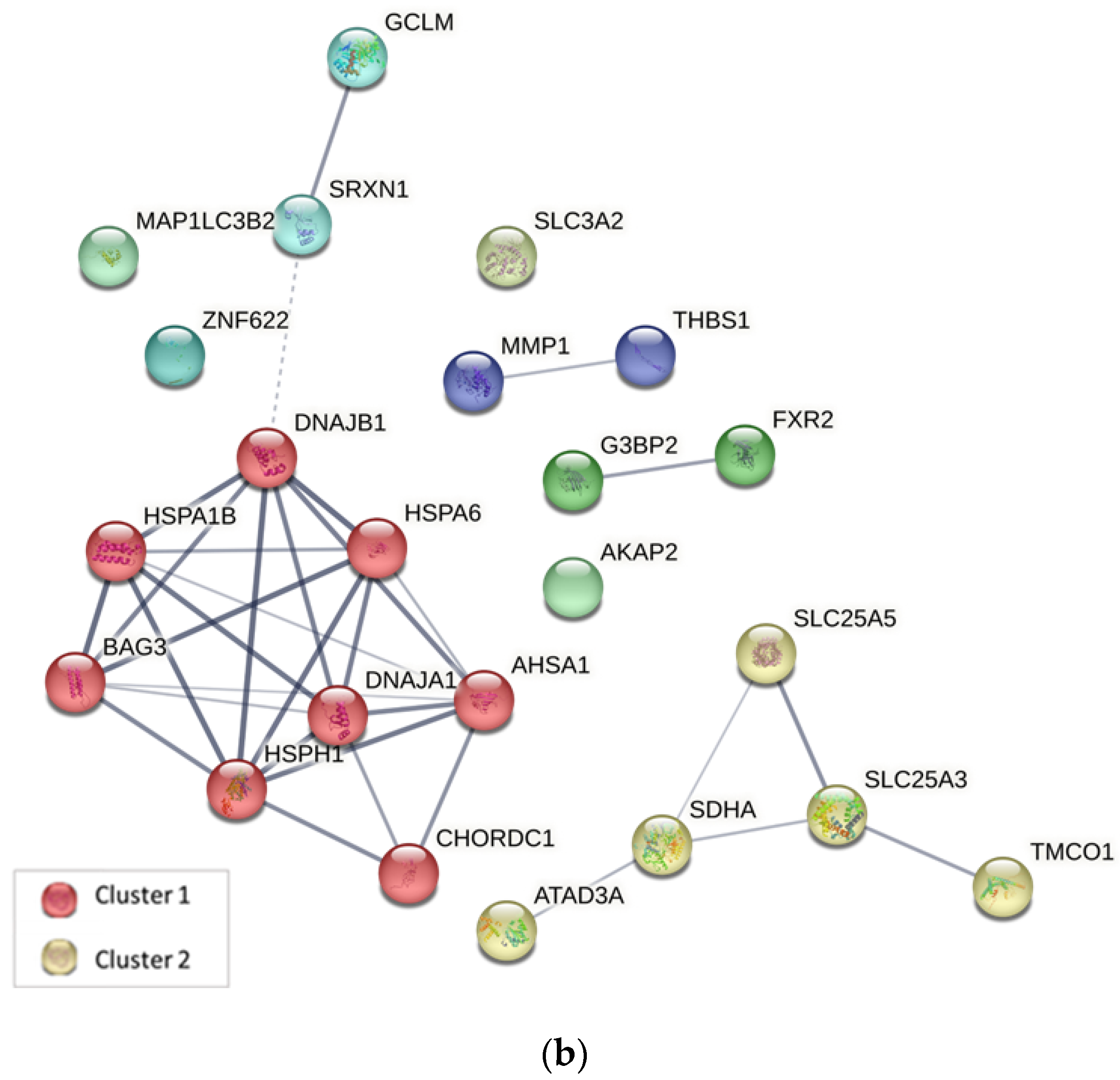
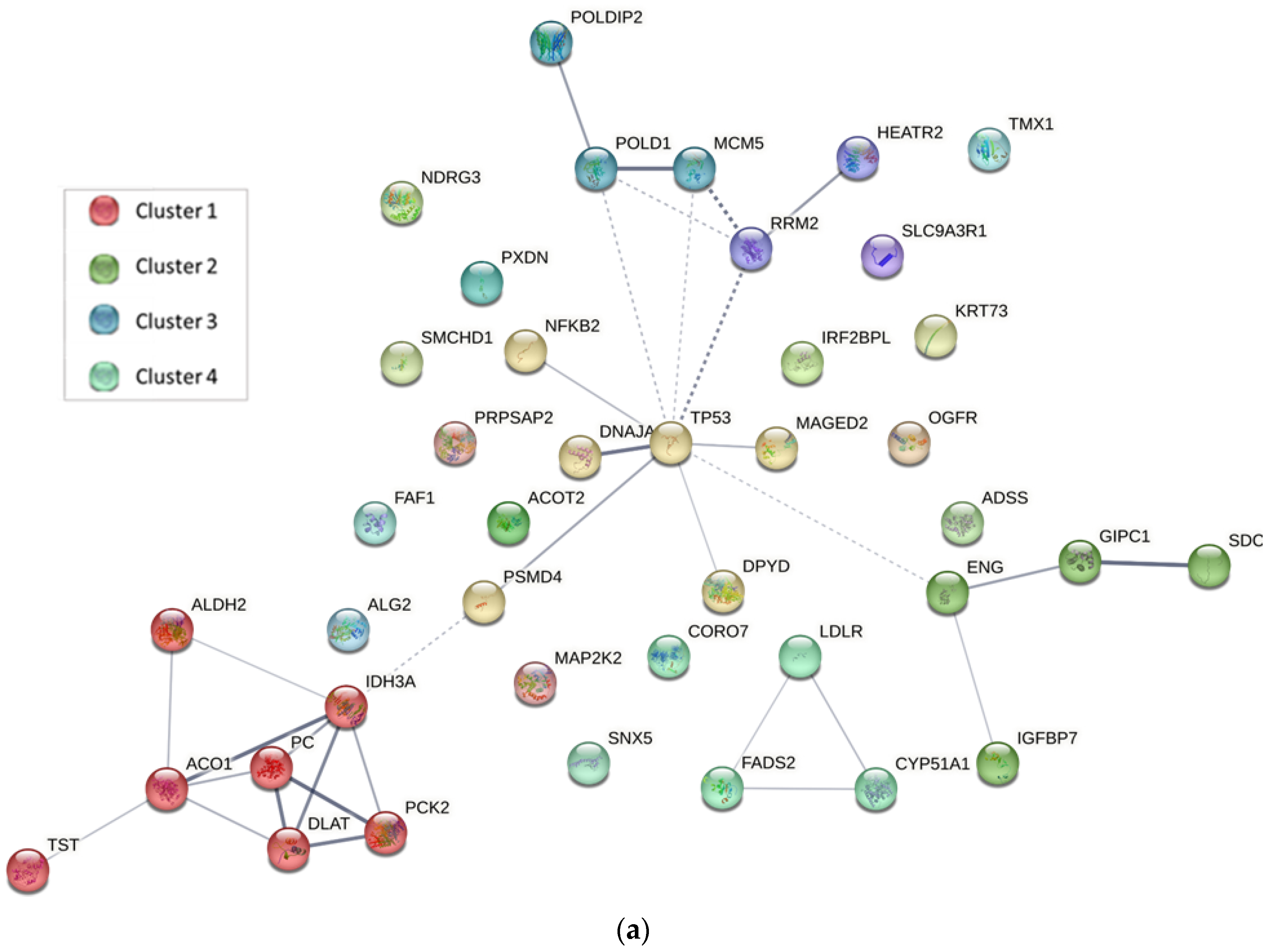
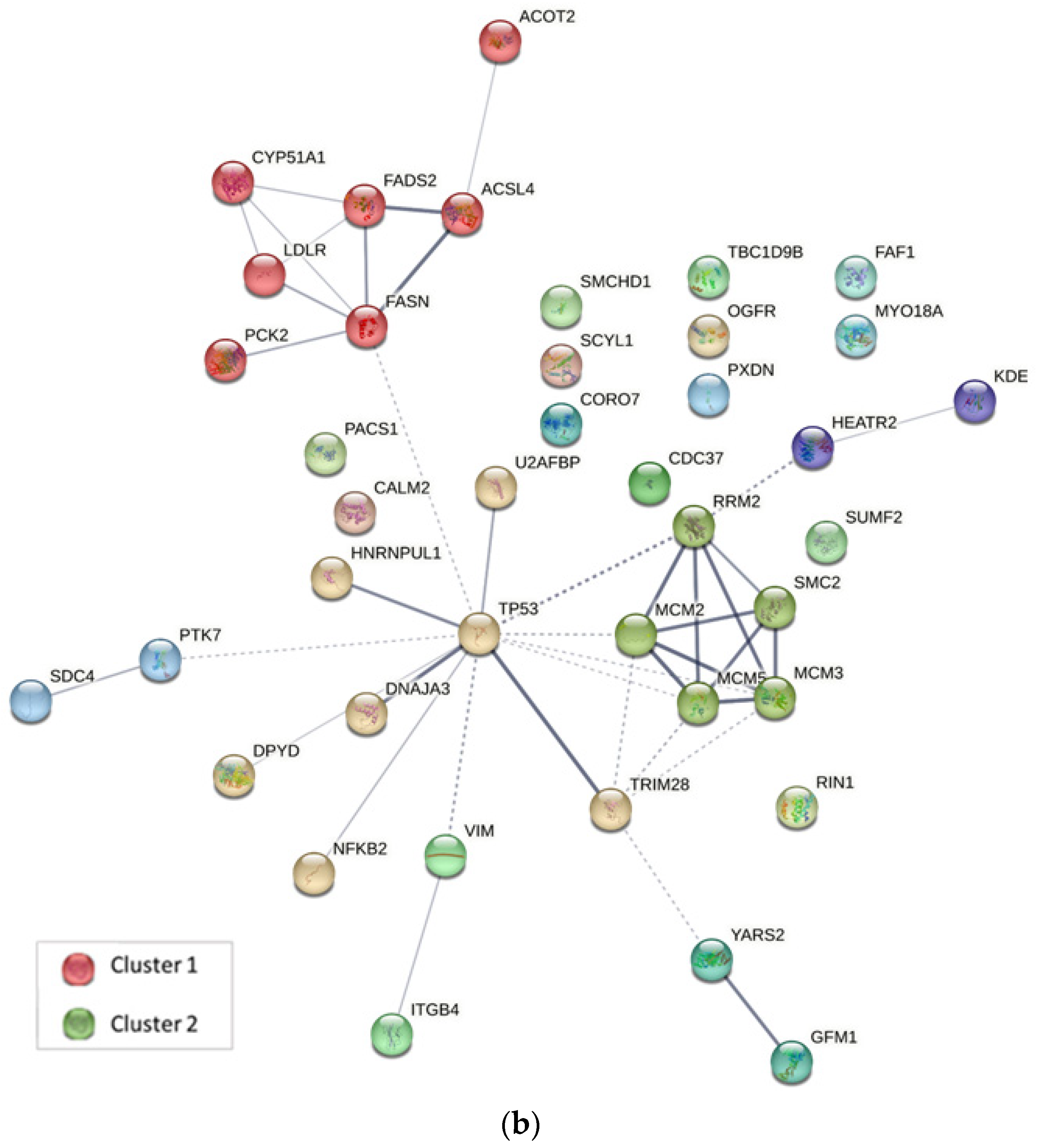
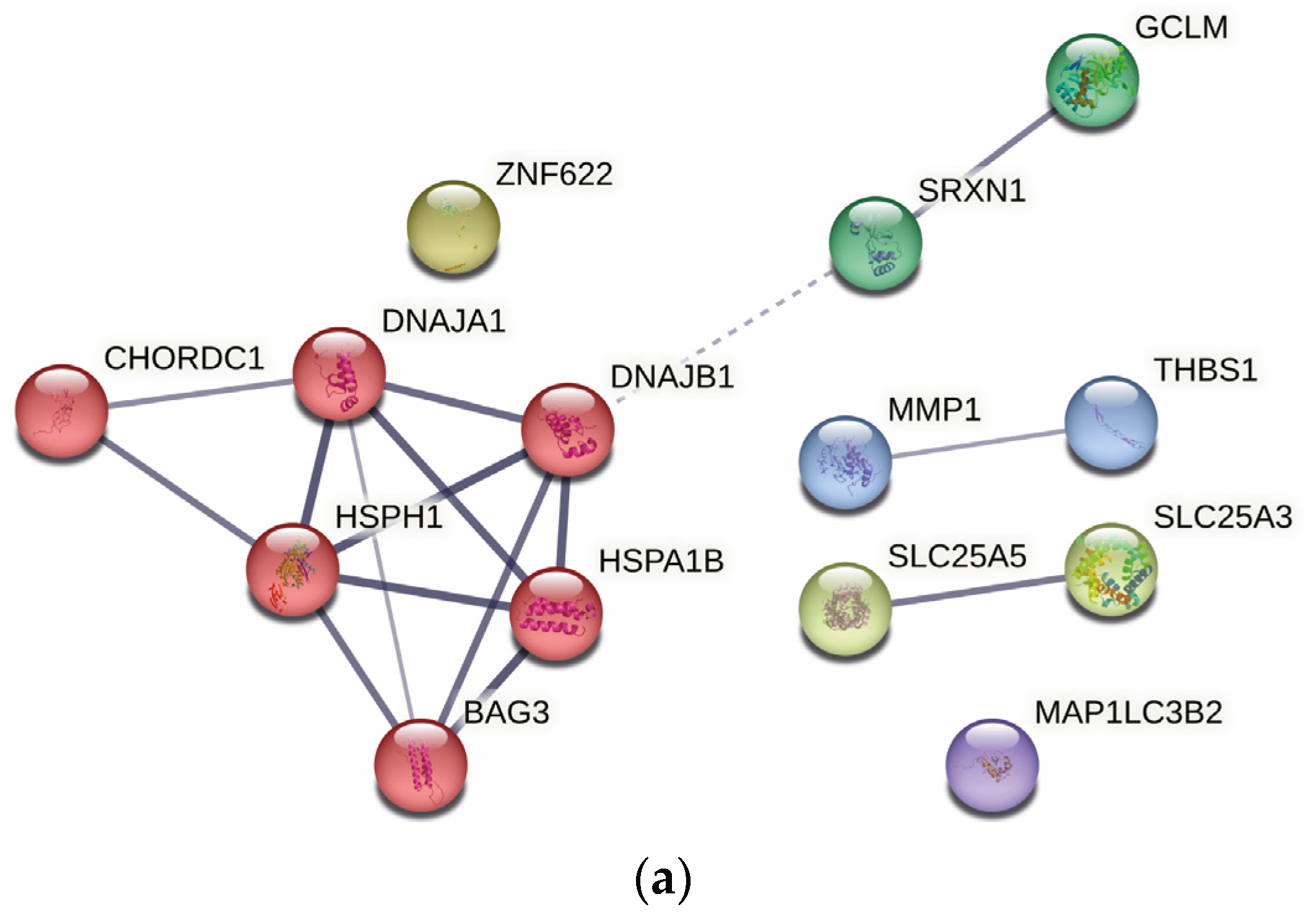
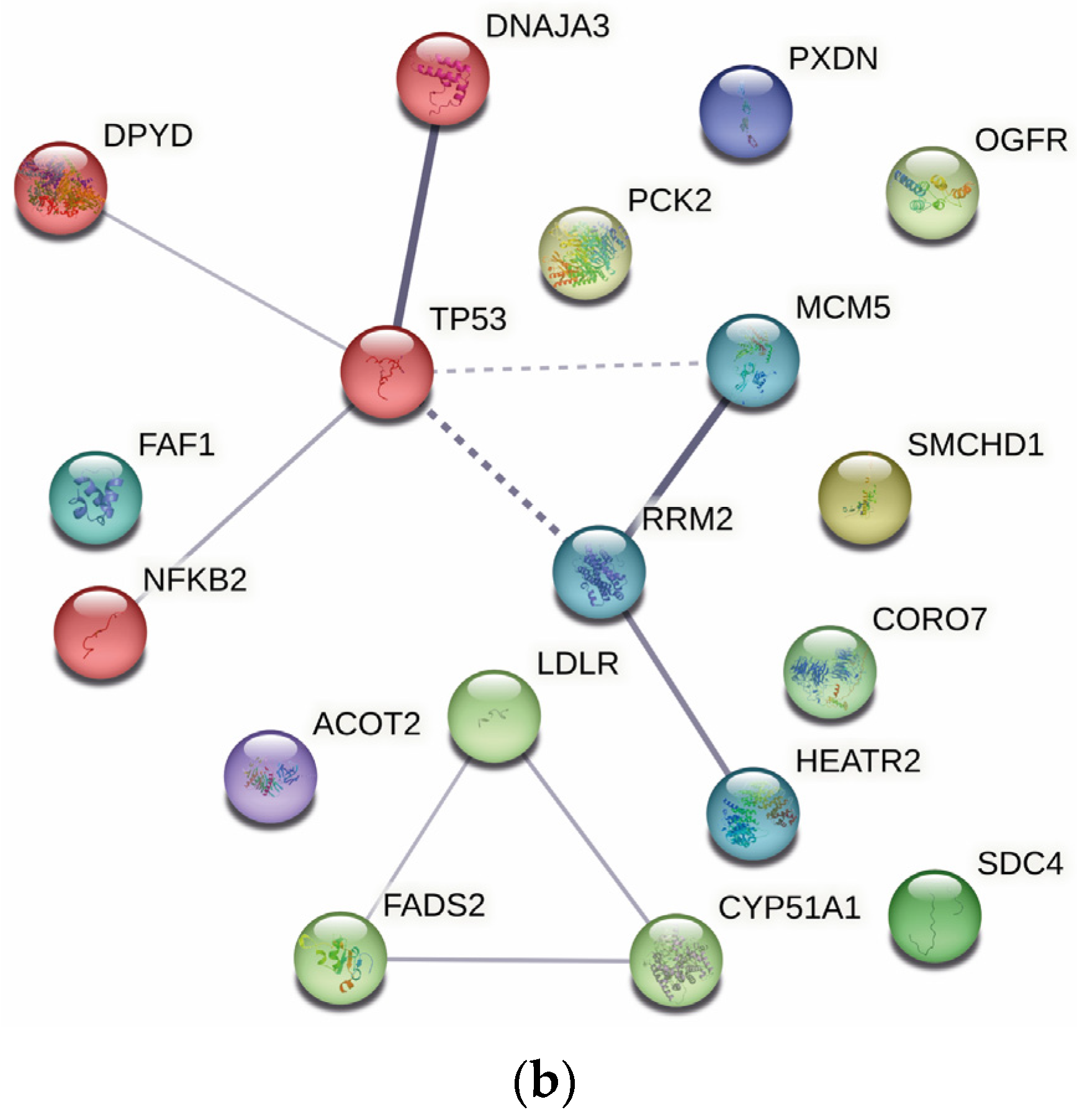
2.4. Protein–Protein Interaction Analysis
2.5. Ingenuity Pathway Analysis
3. Materials and Methods
3.1. Synthesis, Identification, and Preparation of CuHL1 and CuHL2
3.2. Cell Culture
3.3. Protein Sample Preparation
3.4. Protein Identification and Mass Spectrometry
3.5. Bioinformatics Analysis
3.5.1. Functional GO Enrichment Analysis
3.5.2. Protein–Protein Interaction Analysis
3.5.3. Ingenuity Pathway Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, I.O.; Khan, Z. Understanding EGFR Signaling in Breast Cancer and Breast Cancer Stem Cells: Overexpression and Therapeutic Implications. Asian Pac. J. Cancer Prev. 2016, 17, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Manuel, L.; Doolan, M.; Westhuyzen, J.; Shakespeare, T.P.; Aherne, N.J. Effect of Clinical and Treatment Factors on Survival Outcomes of Triple Negative Breast Cancer Patients. Breast Cancer Targets Ther. 2020, 12, 27–35. [Google Scholar] [CrossRef]
- So, J.Y.; Ohm, J.; Lipkowitz, S.; Yang, L. Triple negative breast cancer (TNBC): Non-genetic tumor heterogeneity and immune microenvironment: Emerging treatment options. Pharmacol. Ther. 2022, 237, 108253. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Barry-Holson, K.Q.; Allison, K.H. Breast cancer stem cells: Are we ready to go from bench to bedside? Histopathology 2016, 68, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, M.; Marques, M.P.M.; Ferreira, I.M.P.L.V.O.; Mota-Filipe, H.; Diniz, C. Anticancer activity of palladium-based complexes against triple-negative breast cancer. Drug Discov. Today 2019, 24, 1044–1058. [Google Scholar] [CrossRef]
- Hongthong, K.; Ratanaphan, A. BRCA1-Associated Triple-Negative Breast Cancer and Potential Treatment for Ruthenium-Based Compounds. Curr. Cancer Drug Targets 2016, 16, 606–617. [Google Scholar] [CrossRef]
- Nhukeaw, T.; Temboot, P.; Hansongnern, K.; Ratanaphan, A. Cellular responses of BRCA1-defective and triple-negative breast cancer cells and in vitro BRCA1 interactions induced by metallo-intercalator ruthenium (II) complexes containing chloro-substituted phenylazopyridine. BMC Cancer 2014, 14, 73. [Google Scholar] [CrossRef]
- Boodram, J.N.; Mcgregor, I.J.; Bruno, P.M.; Cressey, P.B.; Hemann, M.T.; Suntharalingam, K. Breast Cancer Stem Cell Potent Copper (II)-Non-Steroidal Anti-Inflammatory Drug Complexes. Angew. Chem.-Int. Ed. 2016, 55, 2845–2850. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: “Copper That Cancer”. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in copper complexes as anticancer agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Balsa, L.M.; Baran, E.J.; León, I.E. Copper complexes as antitumor agents: In vitro and in vivo evidences. Curr. Med. Chem. 2023, 30, 510–557. [Google Scholar] [CrossRef]
- Leon, I.; Cadavid-Vargas, J.; Di Virgilio, A.; Etcheverry, S. Vanadium, Ruthenium and Copper Compounds: A New Class of Nonplatinum Metallodrugs with Anticancer Activity. Curr. Med. Chem. 2016, 24, 112–148. [Google Scholar] [CrossRef]
- Laws, K.; Bineva-Todd, G.; Eskandari, A.; Lu, C.; O’Reilly, N.; Suntharalingam, K. A Copper(II) Phenanthroline Metallopeptide That Targets and Disrupts Mitochondrial Function in Breast Cancer Stem Cells. Angew. Chem.-Int. Ed. 2018, 57, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Balsa, L.M.; Ruiz, M.C.; Santa Maria de la Parra, L.; Baran, E.J.; León, I.E. Anticancer and antimetastatic activity of copper (II)-tropolone complex against human breast cancer cells, breast multicellular spheroids and mammospheres. J. Inorg. Biochem. 2019, 204, 110975. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Palamarciuc, O.; Babak, M.V.; Lim, J.M.; Ohui, K.; Enyedy, E.A.; Shova, S.; Darvasiová, D.; Rapta, P.; Ang, W.H.; et al. Copper (II) thiosemicarbazone complexes induce marked ROS accumulation and promote nrf2-mediated antioxidant response in highly resistant breast cancer cells. Dalt. Trans. 2017, 46, 3833–3847. [Google Scholar] [CrossRef]
- Romo, A.I.B.; Abreu, D.S.; Paulo, T.; Carepo, M.S.P.; Sousa, E.H.S.; Lemus, L.; Aliaga, C.; Batista, A.A.; Nascimento, O.R.; Abruña, H.D.; et al. Hydroxyl Radical Generation and DNA Nuclease Activity: A Mechanistic Study Based on a Surface-Immobilized Copper Thioether Clip-Phen Derivative. Chem. A Eur. J. 2016, 22, 10081–10089. [Google Scholar] [CrossRef]
- Romo, A.I.B.; Carepo, M.P.; Levín, P.; Nascimento, O.R.; Díaz, D.E.; Rodríguez-López, J.; León, I.E.; Bezerra, L.F.; Lemus, L.; Diógenes, I.C.N. Synergy of DNA intercalation and catalytic activity of a copper complex towards improved polymerase inhibition and cancer cell cytotoxicity. Dalt. Trans. 2021, 50, 11931–11940. [Google Scholar] [CrossRef]
- Wang, Y.; Chiu, J.F. Proteomic Approaches in Understanding Action Mechanisms of Metal-Based Anticancer Drugs. Met. Based Drugs 2008, 2008, 716329. [Google Scholar] [CrossRef]
- Rodríguez, M.R.; Balsa, L.M.; Piro, O.E.; Etcheverría, G.A.; García-Tojal, J.; Pis-Diez, R.; León, I.E.; Parajón-Costa, B.P.; González-Baró, A.C. Synthesis, crystal structure, spectroscopic characterization, DFT calculations and cytotoxicity assays of a new cu (II) complex with an acylhydrazone Ligand derived from thiophene. Inorganics 2021, 9, 9. [Google Scholar] [CrossRef]
- Balsa, L.M.; Rodriguez, M.R.; Parajón-Costa, B.S.; González Baró, A.C.; Lavecchia, M.J.; León, I. Anticancer activity and mechanism of action evaluation of an acylhydrazone Cu (II) complex toward breast cancer cells, spheroids and mammospheres. ChemMedChem 2022, 17, e202100520. [Google Scholar] [CrossRef] [PubMed]
- Balsa, L.M.; Ferraresi-Curotto, V.; Lavecchia, M.J.; Echeverría, G.A.; Piro, O.E.; García-Tojal, J.; Pis-Diez, R.; González-Baró, A.C.; León, I.E. Anticancer activity of a new copper (ii) complex with a hydrazone ligand. Structural and spectroscopic characterization, computational simulations and cell mechanistic studies on 2D and 3D breast cancer cell models. Dalt. Trans. 2021, 50, 9812–9826. [Google Scholar] [CrossRef]
- Tiligada, E. Chemotherapy: Induction of stress responses. Endocr. Relat. Cancer 2006, 13, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol.-Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Isella, C.; Medico, E.; Marchiò, L.; Bevilacqua, E.; Hatzoglou, M.; Bussolati, O.; Franchi-Gazzola, R. The thioxotriazole copper (II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells. J. Biol. Chem. 2009, 284, 24306–24319. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The rebel angel: Mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef]
- Hui, L.; Zheng, Y.; Yan, Y.; Bargonetti, J.; Foster, D.A. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 2006, 25, 7305–7310. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad Complex Opposes p63 to Empower TGFβ-Induced Metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Bae, Y.H.; Shin, J.M.; Park, H.J.; Jang, H.O.; Bae, M.K.; Bae, S.K. Gain-of-function mutant p53-R280K mediates survival of breast cancer cells. Genes Genom. 2014, 36, 171–178. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Kumarakulasingham, M.; Rooney, P.H.; Dundas, S.R.; Telfer, C.; Melvin, W.T.; Curran, S.; Murray, G.I. Cytochrome p450 profile of colorectal cancer: Identification of markers of prognosis. Clin. Cancer Res. 2005, 11, 3758–3765. [Google Scholar] [CrossRef]
- Keber, R.; Motaln, H.; Wagner, K.D.; Debeljak, N.; Rassoulzadegan, M.; Ačimovič, J.; Rozman, D.; Horvat, S. Mouse knockout of the cholesterogenic cytochrome p450 lanosterol 14α-demethylase (Cyp51) resembles antley-bixler syndrome. J. Biol. Chem. 2011, 286, 29086–29097. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Botas, J.; Ferruelo, A.J.; Suárez, Y.; Gómez-Coronado, D.; Lasunción, M.A. Induction of apoptosis in p53-null HL-60 cells by inhibition of lanosterol 14-α demethylase. Biochimie 1998, 80, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Mayengbam, S.S.; Singh, A.; Pillai, A.D.; Bhat, M.K. Influence of cholesterol on cancer progression and therapy. Transl. Oncol. 2021, 14, 101043. [Google Scholar] [CrossRef] [PubMed]
- Centonze, G.; Natalini, D.; Piccolantonio, A.; Salemme, V.; Morellato, A.; Arina, P.; Riganti, C.; Defilippi, P. Cholesterol and Its Derivatives: Multifaceted Players in Breast Cancer Progression. Front. Oncol. 2022, 12, 906670. [Google Scholar] [CrossRef]
- Niendorf, A.; Nägele, H.; Gerding, D.; Meyer-Pannwitt, U.; Gebhardt, A. Increased LDL receptor mRNA expression in colon cancer is correlated with a rise in plasma cholesterol levels after curative surgery. Int. J. Cancer 1995, 61, 461–464. [Google Scholar] [CrossRef]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Sebastiano, M.R.; Konstantinidou, G. Targeting long chain acyl-coa synthetases for cancer therapy. Int. J. Mol. Sci. 2019, 20, 3624. [Google Scholar] [CrossRef]
- Cui, Y.; Xing, P.; Wang, Y.; Liu, M.; Qiu, L.; Ying, G.; Li, B. NADPH accumulation is responsible for apoptosis in breast cancer cells induced by fatty acid synthase inhibition. Oncotarget 2017, 8, 32576–32585. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2+ Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Wu, C.H.; Chu, J.S.; Wang, C.K.; Hung, L.F.; Wang, Y.J.; Ho, Y.S.; Chang, J.G.; Lin, S.Y. Involvement of fatty acid-CoA ligase 4 in hepatocellular carcinoma growth: Roles of cyclic AMP and p38 mitogen-activated protein kinase. World J. Gastroenterol. 2005, 11, 2557. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, R.; Cruz-Gil, S.; de Cedrón, M.G.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719. [Google Scholar] [CrossRef]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4 is implicated in drug resistance in breast cancer cell lines involving the regulation of energy-dependent transporter expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q.; Song, J.; Li, S.; Li, X.-L.; Kang, B.; Chen, H.-Y.; Xu, J.-J. Photothermally Triggered Copper Payload Release for Cuproptosis-Promoted Cancer Synergistic Therapy. Angew. Chem. Int. Ed. 2022, 62, e202213922. [Google Scholar] [CrossRef] [PubMed]
- Lei, M. The MCM Complex: Its Role in DNA Replication and Implications for Cancer Therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Guan, Y.; Li, Y.K.; Wu, Q.; Tang, X.J.; Zeng, X.; Ling, H.; Zou, J. The DNA replication regulator MCM6: An emerging cancer biomarker and target. Clin. Chim. Acta 2021, 517, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.K.; Dasilva, S.C.; Bhatta, A.; Rawal, B.; Liu, M.; Korobkova, E.A. Determination of the drug-DNA binding modes using fluorescence-based assays. Anal. Biochem. 2012, 422, 66–73. [Google Scholar] [CrossRef]
- Liu, L.; Mo, J.Y.; Rodriguez-Belmonte, E.M.; Lee, M.Y.W.T. Identification of a fourth subunit of mammalian DNA polymerase delta. J. Biol. Chem. 2000, 275, 18739–18744. [Google Scholar] [CrossRef]
- Kashkin, K.; Chernov, I.; Stukacheva, E.; Monastyrskaya, G.; Uspenskaya, N.; Kopantzev, E.; Sverdlov, E. Cancer specificity of promoters of the genes controlling cell proliferation. J. Cell. Biochem. 2015, 116, 299–309. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, W.; Zhu, X.; Wei, C. p53 inhibits the expression of p125 and the methylation of POLD1 gene promoter by downregulating the Sp1-induced DNMT1 activities in breast cancer. OncoTargets Ther. 2016, 9, 1351–1360. [Google Scholar] [CrossRef]
- Qin, Q.; Tan, Q.; Li, J.; Yang, W.; Lian, B.; Mo, Q.; Wei, C. Elevated expression of POLD1 is associated with poor prognosis in breast cancer. Oncol. Lett. 2018, 16, 5591–5598. [Google Scholar] [CrossRef]
- Liang, Z.J.; Wan, Y.; Zhu, D.D.; Wang, M.X.; Jiang, H.M.; Huang, D.L.; Luo, L.F.; Chen, M.J.; Yang, W.P.; Li, H.M.; et al. Resveratrol Mediates the Apoptosis of Triple Negative Breast Cancer Cells by Reducing POLD1 Expression. Front. Oncol. 2021, 11, 569295. [Google Scholar] [CrossRef]
- Kumar, H.; Kumar, R.M.; Bhattacharjee, D.; Somanna, P.; Jain, V. Role of Nrf2 Signaling Cascade in Breast Cancer: Strategies and Treatment. Front. Pharmacol. 2022, 13, 1357. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Safarzadeh, E.; Safaei, S.; Baradaran, A.; Mohammadi, A.; Hajiasgharzadeh, K.; Derakhshani, A.; Argentiero, A.; Silvestris, N.; Baradaran, B. Current approaches for combination therapy of cancer: The role of immunogenic cell death. Cancers 2020, 12, 1047. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Kaur, P.; Johnson, A.; Northcote-Smith, J.; Lu, C.; Suntharalingam, K. Immunogenic Cell Death of Breast Cancer Stem Cells Induced by an Endoplasmic Reticulum-Targeting Copper (II) Complex. ChemBioChem 2020, 21, 3618–3624. [Google Scholar] [CrossRef]
- Schmitt, F.C.; Longatto Filho, A.; Lopes, J.M. Angiogenesis and Breast Cancer. J. Oncol. 2010, 2010, 576384. [Google Scholar] [CrossRef]
- Ayoub, N.M.; Jaradat, S.K.; Al-Shami, K.M.; Alkhalifa, A.E. Targeting Angiogenesis in Breast Cancer: Current Evidence and Future Perspectives of Novel Anti-Angiogenic Approaches. Front. Pharmacol. 2022, 13, 485. [Google Scholar] [CrossRef]
- Shao, J.; Liu, X.; Zhu, L.; Yen, Y. Targeting ribonucleotide reductase for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1423–1437. [Google Scholar] [CrossRef]
- Zhuang, S.; Li, L.; Zang, Y.; Li, G.; Wang, F. RRM2 elicits the metastatic potential of breast cancer cells by regulating cell invasion, migration and VEGF expression via the PI3K/AKT signaling. Oncol. Lett. 2020, 19, 3349–3355. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Fold-Change | p-Value |
|---|---|---|---|
| HSPA1B | Heat shock 70 kDa protein 1B | 11.9067 | 0.0000 |
| SRXN1 | Sulfiredoxin-1 | 4.8384 | 0.0144 |
| CSTF3 | Cleavage stimulation factor subunit 3 | 4.3157 | 0.0128 |
| THBS1 | Thrombospondin-1 | 3.9412 | 0.0016 |
| CHORDC1 | Cysteine and histidine-rich domain-containing protein 1 | 3.8146 | 0.0028 |
| BAG3 | BAG family molecular chaperone regulator 3 | 3.7564 | 0.0058 |
| SLC25A3 | Phosphate carrier protein, mitochondrial | 3.6397 | 0.0046 |
| HSPH1 | Heat shock protein 105 kDa | 3.5744 | 0.0007 |
| DNAJB1 | DnaJ homolog subfamily B member 1 | 3.5735 | 0.0000 |
| GCLM | Glutamate–cysteine ligase regulatory subunit | 3.4930 | 0.0005 |
| SLC25A5 | ADP/ATP translocase 2 | 3.2590 | 0.0085 |
| ARF3 | ADP-ribosylation factor 3 | 2.8776 | 0.0335 |
| MAP1LC3B2 | Microtubule-associated proteins 1A/1B light chain 3 beta 2 | 2.7896 | 0.0222 |
| DDX21 | Nucleolar RNA helicase 2 | 2.7624 | 0.0244 |
| KTN1 | Kinectin | 2.6451 | 0.0302 |
| MMP1 | Interstitial collagenase | 2.6260 | 0.0060 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 2.5367 | 0.0247 |
| FUS | RNA-binding protein FUS | 2.3718 | 0.0305 |
| DNAJA1 | DnaJ homolog subfamily A member 1 | 2.2815 | 0.0003 |
| TECR | Very-long-chain enoyl-CoA reductase | 2.2753 | 0.0422 |
| ARHGEF18 | Rho guanine nucleotide exchange factor 18 | 2.1868 | 0.0453 |
| RPL12 | 60S ribosomal protein L12 | 2.1825 | 0.0442 |
| ZNF622 | Zinc finger protein 622 | 2.1629 | 0.0360 |
| RPL21 | 60S ribosomal protein L21 | 2.0765 | 0.0283 |
| TOP2A | DNA topoisomerase 2-alpha | 2.0762 | 0.0075 |
| RPS8 | 40S ribosomal protein S8 | 2.0729 | 0.0227 |
| RPL7A | 60S ribosomal protein L7a | 2.0663 | 0.0055 |
| RPL10 | 60S ribosomal protein L10 | 2.0453 | 0.0303 |
| SDC4 | Syndecan-4 | −54.1800 | 0.0332 |
| CYP51A1 | Lanosterol 14-alpha demethylase | −11.7752 | 0.0010 |
| FADS2 | Fatty acid-desaturase | −8.9256 | 0.0007 |
| ALG2 | Alpha-1,3/1,6-mannosyltransferase ALG2 | −8.8383 | 0.0036 |
| RRM2 | Ribonucleoside-diphosphate reductase subunit M2 | −5.7327 | 0.0040 |
| SMCHD1 | Structural maintenance of chromosomes flexible hinge domain-containing protein 1 | −5.6365 | 0.0302 |
| DPYD | Dihydropyrimidine dehydrogenase [NADP(+)] | −4.8430 | 0.0003 |
| POLD1 | DNA polymerase | −4.4882 | 0.0165 |
| NFKB2 | Nuclear factor NF-kappa-B p100 subunit | −4.2297 | 0.0051 |
| IRF2BPL | Probable E3 ubiquitin-protein ligase IRF2BPL | −3.9927 | 0.0064 |
| TP53 | Cellular tumor antigen p53 | −3.4861 | 0.0015 |
| LDLR | Low-density lipoprotein receptor (Fragment) | −3.3732 | 0.0065 |
| TMX1 | Thioredoxin-related transmembrane protein 1 | −3.3412 | 0.0015 |
| PSMD4 | 26S proteasome non-ATPase regulatory subunit 4 | −3.3016 | 0.0123 |
| DNAAF5 | Dynein axonemal assembly factor 5 | −3.0889 | 0.0065 |
| TST | Thiosulfate sulfurtransferase | −3.0300 | 0.0075 |
| PXDN | Peroxidasin homolog | −3.0016 | 0.0003 |
| OGFR | Opioid growth factor receptor | −2.9898 | 0.0177 |
| ENG | Endoglin | −2.7407 | 0.0145 |
| FAF1 | FAS-associated factor 1 | −2.5783 | 0.0183 |
| MCM5 | DNA replication licensing factor MCM5 | −2.5649 | 0.0022 |
| ADSS2 | Adenylosuccinate synthetase isozyme 2 | −2.4898 | 0.0118 |
| GIPC1 | PDZ domain-containing protein GIPC1 | −2.4031 | 0.0494 |
| ACO1 | Cytoplasmic aconitate hydratase | −2.3932 | 0.0013 |
| PRPSAP2 | Phosphoribosyl pyrophosphate synthase-associated protein 2 | −2.3817 | 0.0099 |
| DNAJA3 | DnaJ homolog subfamily A member 3, mitochondrial | −2.3163 | 0.0022 |
| DLAT | Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial | −2.3145 | 0.0009 |
| SNX5 | Sorting nexin-5 | −2.2927 | 0.0082 |
| PCK2 | Phosphoenolpyruvate carboxykinase [GTP], mitochondrial | −2.2247 | 0.0184 |
| IGFBP7 | Insulin-like growth factor-binding protein 7 | −2.2145 | 0.0077 |
| NDRG3 | N-myc downstream-regulated gene 3 protein | −2.1762 | 0.0005 |
| KRT73 | Keratin, type II cytoskeletal 73 | −2.1747 | 0.0458 |
| PC | Pyruvate carboxylase, mitochondrial | −2.1747 | 0.0187 |
| CORO7 | Coronin | −2.1529 | 0.0035 |
| POLDIP2 | Polymerase delta-interacting protein 2 | −2.1529 | 0.0011 |
| ACOT2 | Acyl-coenzyme A thioesterase 2, mitochondrial | −2.1328 | 0.0021 |
| MAP2K2 | Dual-specificity mitogen-activated protein kinase kinase 2 | −2.1182 | 0.0009 |
| MAGED2 | Melanoma-associated antigen D2 | −2.1069 | 0.0476 |
| IDH3A | Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial | −2.0820 | 0.0012 |
| ALDH2 | Aldehyde dehydrogenase, mitochondrial | −2.0759 | 0.0003 |
| SLC9A3R1 | Na(+)/H(+) exchange regulatory cofactor NHE-RF1 | −2.0756 | 0.0071 |
| Gene | Protein | Fold-Change | p-Value |
|---|---|---|---|
| HSPA6 | Heat shock 70 kDa protein 6 | 135.1126 | 0.0001 |
| HSPA1B | Heat shock 70 kDa protein 1B | 12.4443 | 0.0000 |
| SRXN1 | Sulfiredoxin-1 | 4.7883 | 0.0157 |
| BAG3 | BAG family molecular chaperone regulator 3 | 4.4887 | 0.0045 |
| G3BP2 | Ras GTPase-activating protein-binding protein 2 | 3.6714 | 0.0310 |
| THBS1 | Thrombospondin-1 | 3.5660 | 0.0120 |
| CHORDC1 | Cysteine and histidine-rich domain-containing protein 1 | 3.5336 | 0.0057 |
| FXR2 | Fragile X mental retardation syndrome-related protein 2 | 3.4133 | 0.0198 |
| SLC25A3 | Phosphate carrier protein, mitochondrial | 3.2324 | 0.0178 |
| DNAJB1 | DnaJ homolog subfamily B member 1 | 3.0959 | 0.0000 |
| SLC25A5 | ADP/ATP translocase 2 | 3.0855 | 0.0253 |
| GCLM | Glutamate–cysteine ligase regulatory subunit | 2.8046 | 0.0024 |
| ATAD3A | ATPase family AAA domain-containing protein 3A | 2.7501 | 0.0031 |
| MMP1 | Interstitial collagenase | 2.7465 | 0.0051 |
| MAP1LC3B2 | Microtubule-associated proteins 1A/1B light chain 3 beta 2 | 2.7147 | 0.0338 |
| HSPH1 | Heat shock protein 105 kDa | 2.7037 | 0.0038 |
| SDHA | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | 2.2722 | 0.0472 |
| DNAJA1 | DnaJ homolog subfamily A member 1 | 2.2136 | 0.0003 |
| ZNF622 | Zinc finger protein 622 | 2.1139 | 0.0070 |
| AKAP2 | A-kinase anchor protein 2 | 2.0990 | 0.0117 |
| TMCO1 | Calcium-load-activated calcium channel | 2.0869 | 0.0164 |
| SLC3A2 | 4F2 cell-surface antigen heavy chain | 2.0386 | 0.0031 |
| AHSA1 | Activator of 90 kDa heat shock protein ATPase homolog 1 | 1.9963 | 0.0228 |
| SMCHD1 | Structural maintenance of chromosomes flexible hinge domain-containing protein 1 | −8.2820 | 0.0014 |
| NFKB2 | Nuclear factor NF-kappa-B p100 subunit | −5.8093 | 0.0379 |
| DPYD | Dihydropyrimidine dehydrogenase [NADP(+)] | −5.6316 | 0.0001 |
| RRM2 | Ribonucleoside-diphosphate reductase subunit M2 | −5.2034 | 0.0018 |
| FADS2 | Acyl-CoA 6-desaturase | −5.0562 | 0.0169 |
| DNAAF5 | Dynein axonemal assembly factor 5 | −4.3873 | 0.0202 |
| U2AF1 | Splicing factor U2AF 35 kDa subunit | −4.2363 | 0.0027 |
| SDC4 | Syndecan-4 | −4.2206 | 0.0232 |
| ACSL4 | Long-chain-fatty-acid--CoA ligase 4 | −3.7610 | 0.0026 |
| CYP51A1 | Lanosterol 14-alpha demethylase | −3.6858 | 0.0032 |
| TP53 | Cellular tumor antigen p53 | −3.6284 | 0.0005 |
| GFM1 | Elongation factor G, mitochondrial | −3.4629 | 0.0032 |
| LDLR | Low-density lipoprotein receptor | −3.2341 | 0.0284 |
| OGFR | Opioid growth factor receptor OS = Homo sapiens | −3.2299 | 0.0139 |
| RIN1 | Ras and Rab interactor 1 | −2.7981 | 0.0113 |
| FAF1 | FAS-associated factor 1 | −2.6790 | 0.0022 |
| MCM2 | DNA replication licensing factor MCM2 | −2.6450 | 0.0239 |
| PCK2 | Phosphoenolpyruvate carboxykinase [GTP], mitochondrial | −2.6115 | 0.0004 |
| SUMF2 | Inactive C-alpha-formylglycine-generating enzyme 2 | −2.5908 | 0.0432 |
| HNRNPUL1 | Heterogeneous nuclear ribonucleoprotein U-like protein 1 | −2.5669 | 0.0440 |
| PXDN | Peroxidasin homolog | −2.5552 | 0.0031 |
| MCM3 | DNA replication licensing factor MCM3 | −2.5299 | 0.0332 |
| DNAJA3 | DnaJ homolog subfamily A member 3, mitochondrial | −2.5193 | 0.0231 |
| PACS1 | Phosphofurin acidic cluster sorting protein 1 | −2.4865 | 0.0405 |
| POGLUT3 | Protein O-glucosyltransferase 3 | −2.3599 | 0.0119 |
| SCYL1 | N-terminal kinase-like protein | −2.3123 | 0.0020 |
| FASN | Fatty acid synthase | −2.2785 | 0.0382 |
| VIM | Vimentin | −2.2682 | 0.0343 |
| MCM5 | DNA replication licensing factor MCM5 | −2.2670 | 0.0021 |
| TRIM28 | Transcription intermediary factor 1-beta | −2.2658 | 0.0431 |
| CDC37 | Hsp90 co-chaperone Cdc37 | −2.2547 | 0.0236 |
| YARS2 | Tyrosine--tRNA ligase, mitochondrial | −2.2501 | 0.0139 |
| SMC2 | Structural maintenance of chromosomes protein 2 | −2.2447 | 0.0288 |
| ITGB4 | Integrin beta-4 | −2.2163 | 0.0362 |
| PTK7 | Inactive tyrosine-protein kinase 7 | −2.1469 | 0.0128 |
| CORO7 | Coronin | −2.1154 | 0.0290 |
| ACOT2 | Acyl-coenzyme A thioesterase 2, mitochondrial | −2.1016 | 0.0002 |
| TBC1D9B | TBC1 domain family member 9B | −2.0811 | 0.0103 |
| MYO18A | Unconventional myosin-XVIIIa | −2.0581 | 0.0201 |
| CALM2 | Calmodulin-2 | −2.0396 | 0.0306 |
| Canonical Pathways | p-Value | Proteins |
|---|---|---|
| EIF2 Signaling | 4.78 × 10−6 | ACTC1, MAP2K2, RPL10, RPL12, RPL21, RPL7A, RPS8 |
| Unfolded protein response | 6.97 × 10−6 | DNAJA1, DNAJA3, DNAJB1, HSPA1A/HSPA1B, HSPH1 |
| Induction of Apoptosis by HIV1 | 4.09 × 10−5 | NFKB2, SLC25A3, SLC25A5, TP53 |
| NRF2-mediated Oxidative Stress Response | 7.13 × 10−5 | ACTC1, DNAJA1, DNAJA3, DNAJB1, GCLM, MAP2K2 |
| BAG2 Signaling Pathway | 1.12 × 10−4 | HSPA1A/HSPA1B, NFKB2, PSMD4, TP53 |
| Aldosterone Signaling in Epithelial Cells | 1.39 × 10−4 | DNAJA1, DNAJB1, HSPA1A/HSPA1B, HSPH1, MAP2K2 |
| Bladder Cancer Signaling | 3.87 × 10−4 | MAP2K2, MMP1, THBS1, TP53 |
| Cell Cycle Control of Chromosomal Replication | 6.08 × 10−4 | MCM5, POLD1, TOP2A |
| Protein Ubiquitination Pathway | 1.30 × 10−3 | DNAJA1, DNAJB1, HSPA1A/HSPA1B, HSPH1, PSMD4 |
| Ribonucleotide Reductase Signaling Pathway | 1.61 × 10−3 | NFKB2, RRM2, THBS1, TP53 |
| HIF1α Signaling | 3.34 × 10−3 | HSPA1A/HSPA1B, MAP2K2, MMP1, TP53 |
| Apoptosis Signaling | 3.61 × 10−3 | MAP2K2, NFKB2, TP53 |
| Autophagy | 3.82 × 10−3 | MAP1LC3B2, MAP2K2, NFKB2, TP53 |
| HER-2 Signaling in Breast Cancer | 4.56 × 10−3 | ARF3, MAP2K2, NFKB2, TP53 |
| Canonical Pathways | p-Value | Proteins |
|---|---|---|
| Unfolded protein response | 1.28 × 10−7 | DNAJA1, DNAJA3, DNAJB1, HSPA6, HSPA1A/HSPA1B, HSPH1 |
| Sirtuin Signaling Pathway | 1.10 × 10−5 | MAP1LC3B2, NFKB2, PCK2, SDHA, SLC25A5, TP53, TRIM28 |
| Induction of Apoptosis by HIV1 | 2.51 × 10−5 | NFKB2, SLC25A3, SLC25A5, TP53 |
| BAG2 Signaling Pathway | 6.90 × 10−5 | HSPA6, HSPA1A/HSPA1B, NFKB2, TP53 |
| Aldosterone Signaling in Epithelial Cells | 7.73 × 10−5 | DNAJA1, DNAJB1, HSPA6, HSPA1A/HSPA1B, HASPH1 |
| HIF1α Signaling | 2.10 × 10−4 | HSPA6, HSPA1A/HSPA1B, MMP1, TP53, VIM |
| Autophagy | 2.50 × 10−4 | CALM1, MAP1LC3B2, NFKB2, SLC3A2, TP53 |
| LXR/RXR Activation | 3.01 × 10−4 | CYP51A1, FASN, LDLR, NFKB2 |
| Cell Cycle Control of Chromosomal Replication | 4.24 × 10−4 | MCM2, MCM3, MCM5 |
| Ribonucleotide Reductase Signaling Pathway | 1.02 × 10−3 | NFKB2, RRM2, THBS1, TP53 |
| Immunogenic Cell Death Signaling Pathway | 1.69 × 10−3 | HSPA6, HSPA1A/HSPA1B, NFKB2 |
| PI3K/AKT Signaling | 1.85 × 10−3 | CDC37, ITGB4, NFKB2, TP53 |
| NRF2-mediated Oxidative Stress Response | 3.41 × 10−3 | DNAJA1, DNAJA3, DNAJB1, GCLM |
| Inhibition of Angiogenesis by TSP1 | 3.55 × 10−3 | THBS1, TP53 |
| MYC-Mediated Apoptosis Signaling | 7.56 × 10−3 | NFKB2, TP53 |
| FAT10 Cancer Signaling Pathway | 7.56 × 10−3 | NFKB2, TP53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balsa, L.M.; Rodriguez, M.R.; Ferraresi-Curotto, V.; Parajón-Costa, B.S.; Gonzalez-Baró, A.C.; León, I.E. Finding New Molecular Targets of Two Copper(II)-Hydrazone Complexes on Triple-Negative Breast Cancer Cells Using Mass-Spectrometry-Based Quantitative Proteomics. Int. J. Mol. Sci. 2023, 24, 7531. https://doi.org/10.3390/ijms24087531
Balsa LM, Rodriguez MR, Ferraresi-Curotto V, Parajón-Costa BS, Gonzalez-Baró AC, León IE. Finding New Molecular Targets of Two Copper(II)-Hydrazone Complexes on Triple-Negative Breast Cancer Cells Using Mass-Spectrometry-Based Quantitative Proteomics. International Journal of Molecular Sciences. 2023; 24(8):7531. https://doi.org/10.3390/ijms24087531
Chicago/Turabian StyleBalsa, Lucia M., María R. Rodriguez, Verónica Ferraresi-Curotto, Beatriz S. Parajón-Costa, Ana C. Gonzalez-Baró, and Ignacio E. León. 2023. "Finding New Molecular Targets of Two Copper(II)-Hydrazone Complexes on Triple-Negative Breast Cancer Cells Using Mass-Spectrometry-Based Quantitative Proteomics" International Journal of Molecular Sciences 24, no. 8: 7531. https://doi.org/10.3390/ijms24087531
APA StyleBalsa, L. M., Rodriguez, M. R., Ferraresi-Curotto, V., Parajón-Costa, B. S., Gonzalez-Baró, A. C., & León, I. E. (2023). Finding New Molecular Targets of Two Copper(II)-Hydrazone Complexes on Triple-Negative Breast Cancer Cells Using Mass-Spectrometry-Based Quantitative Proteomics. International Journal of Molecular Sciences, 24(8), 7531. https://doi.org/10.3390/ijms24087531