
SAA1 Has Potential as a Prognostic Biomarker Correlated with Cell Proliferation, Migration, and an Indicator for Immune Infiltration of Tumor Microenvironment in Clear Cell Renal Cell Carcinoma


Abstract
1. Introduction
2. Results
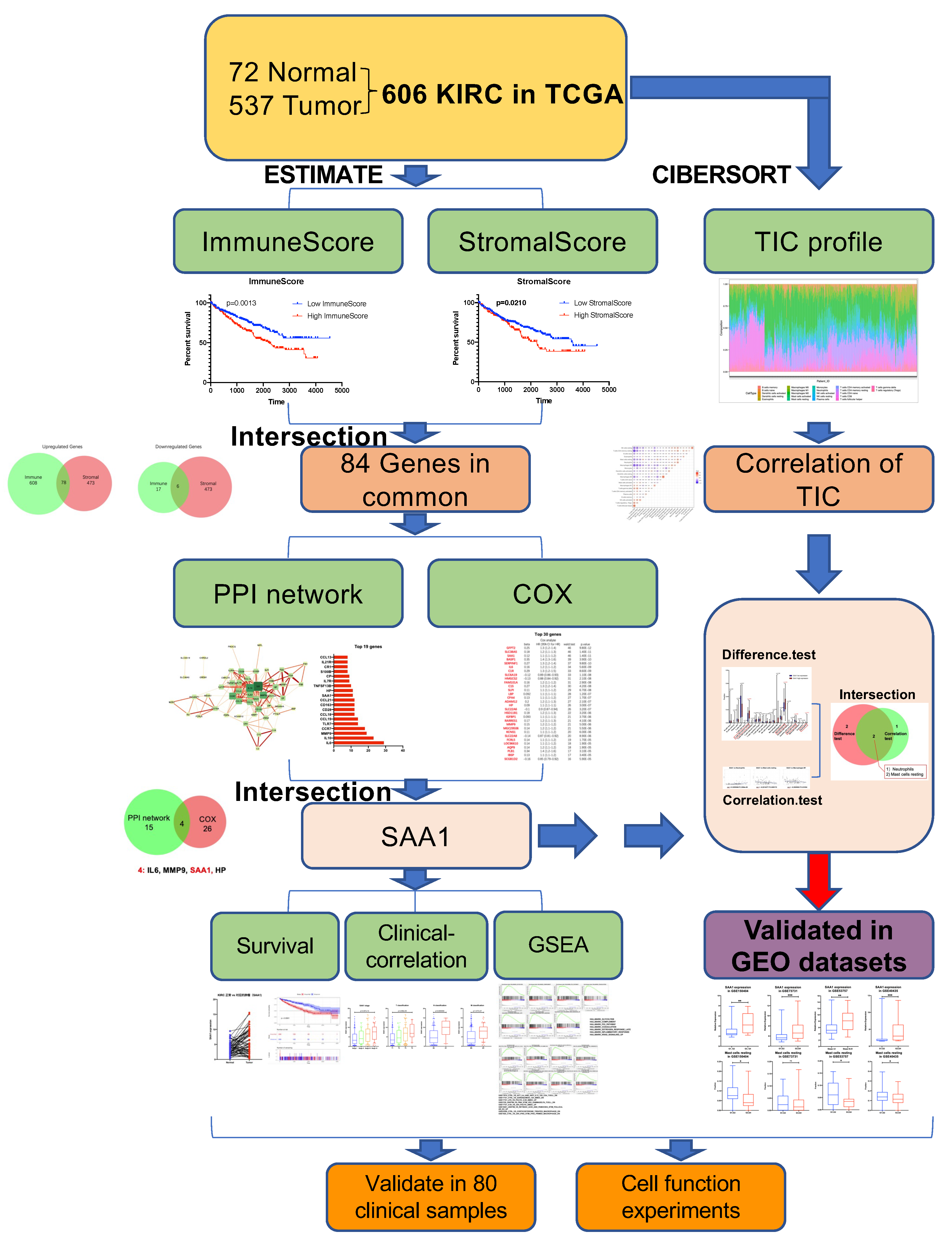
2.1. Data Acquisition and Analysis Workflow of This Study
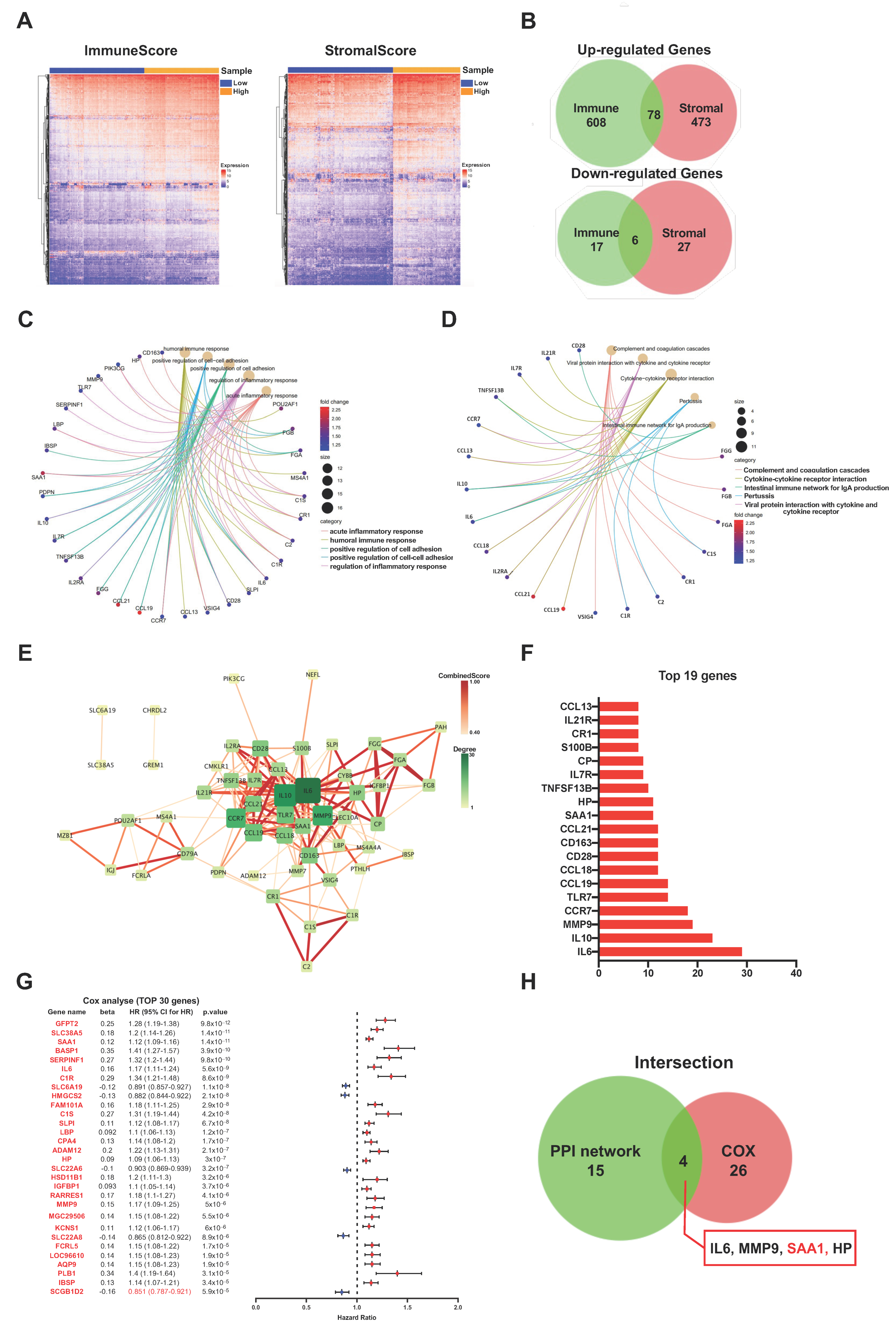
2.2. ImmuneScore, StromalScore, and ESTIMATEScore Are Significantly Associated with Survival and Clinical Features of ccRCC
2.3. The Common DEGs of ImmuneScore and StromalScore Were Mainly Regarded as Immune-Related Genes
2.4. PPI Network Analysis, Univariate Cox Regression of 84 DEGs
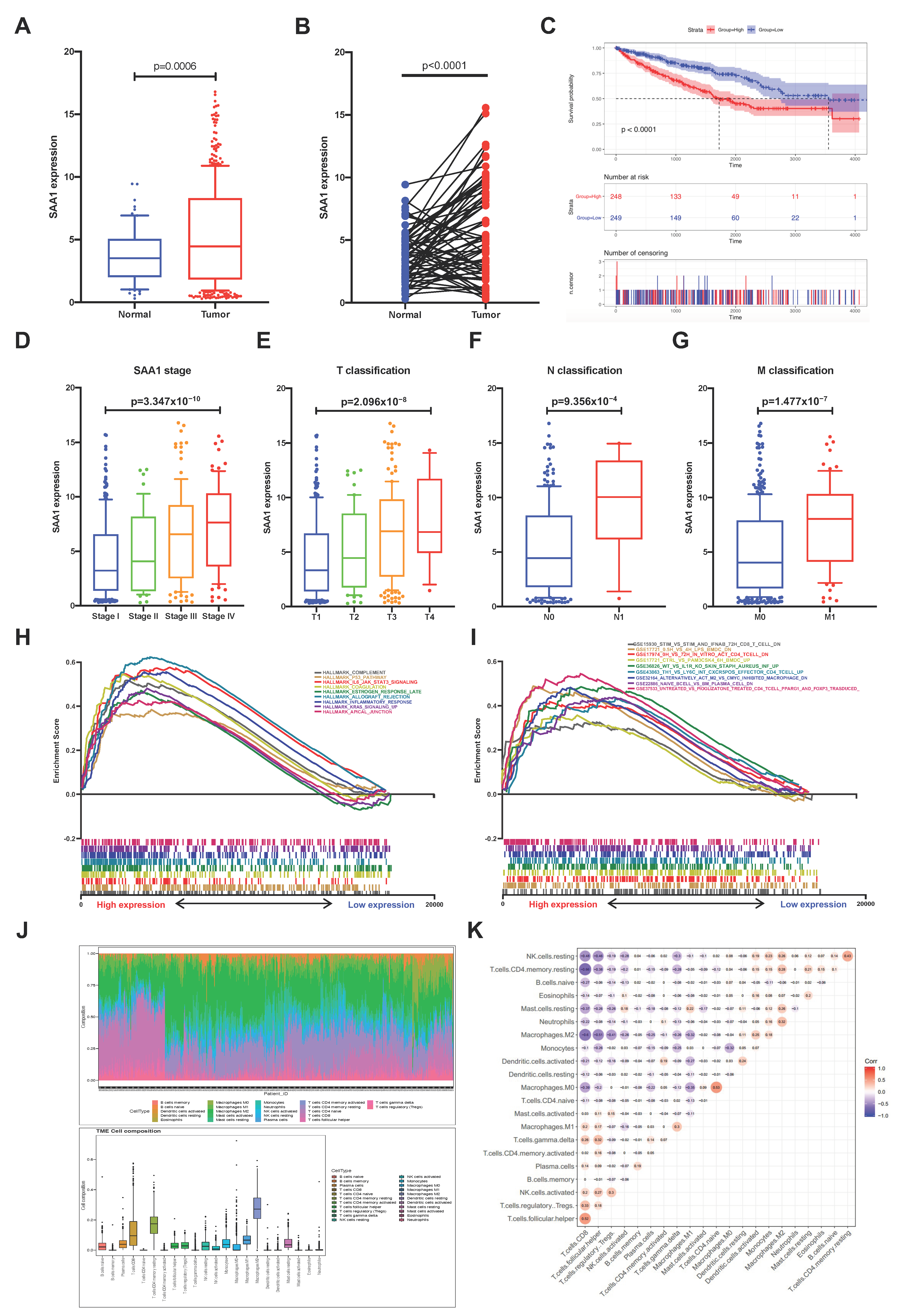
2.5. The Correlation of SAA1 with Survival and TNM Stages of ccRCC Patients
2.6. SAA1 Can Be an Indicator of TME Status for ccRCC Patients
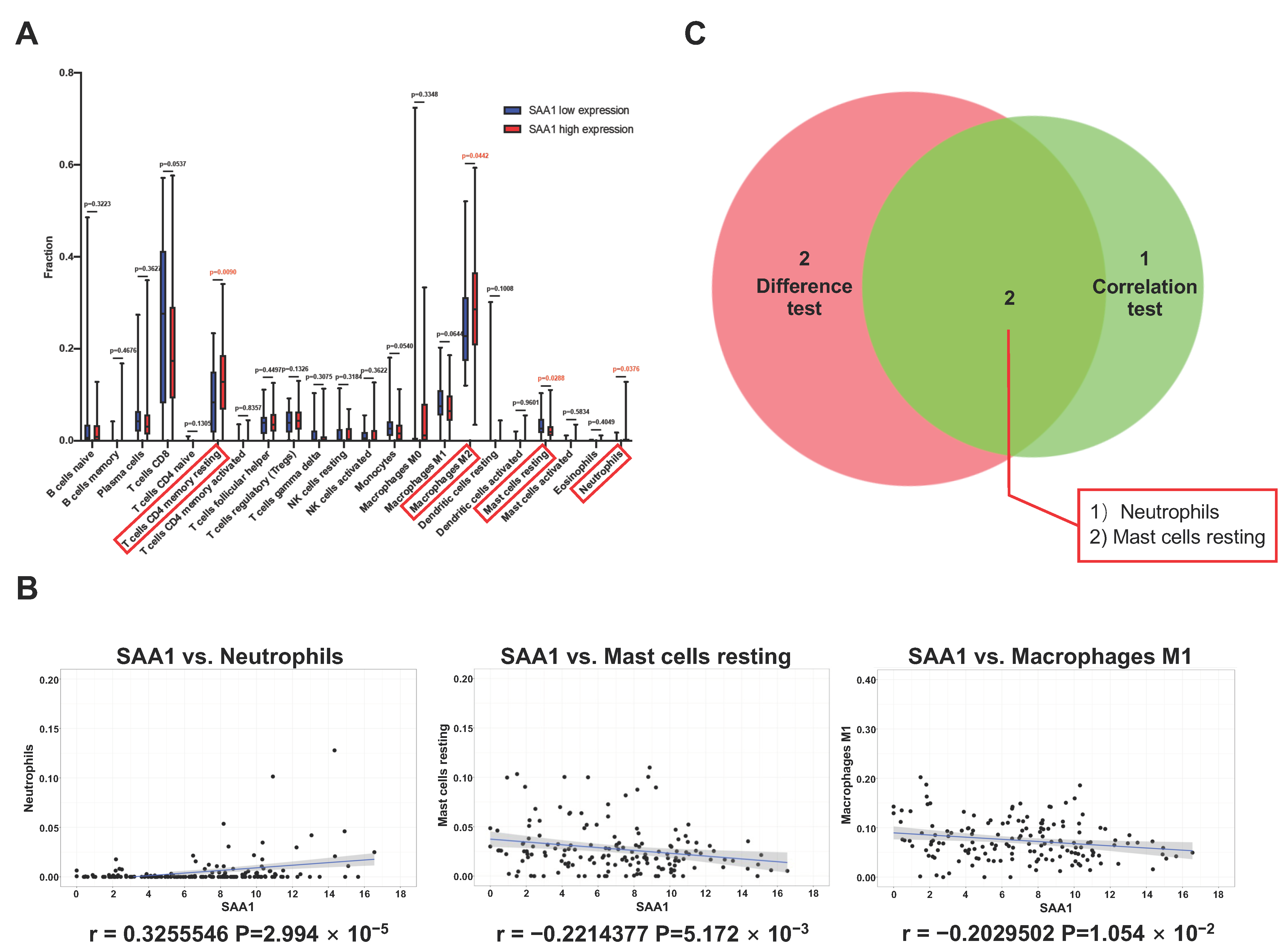
2.7. Correlation between SAA1 Expression and Proportion of TICs
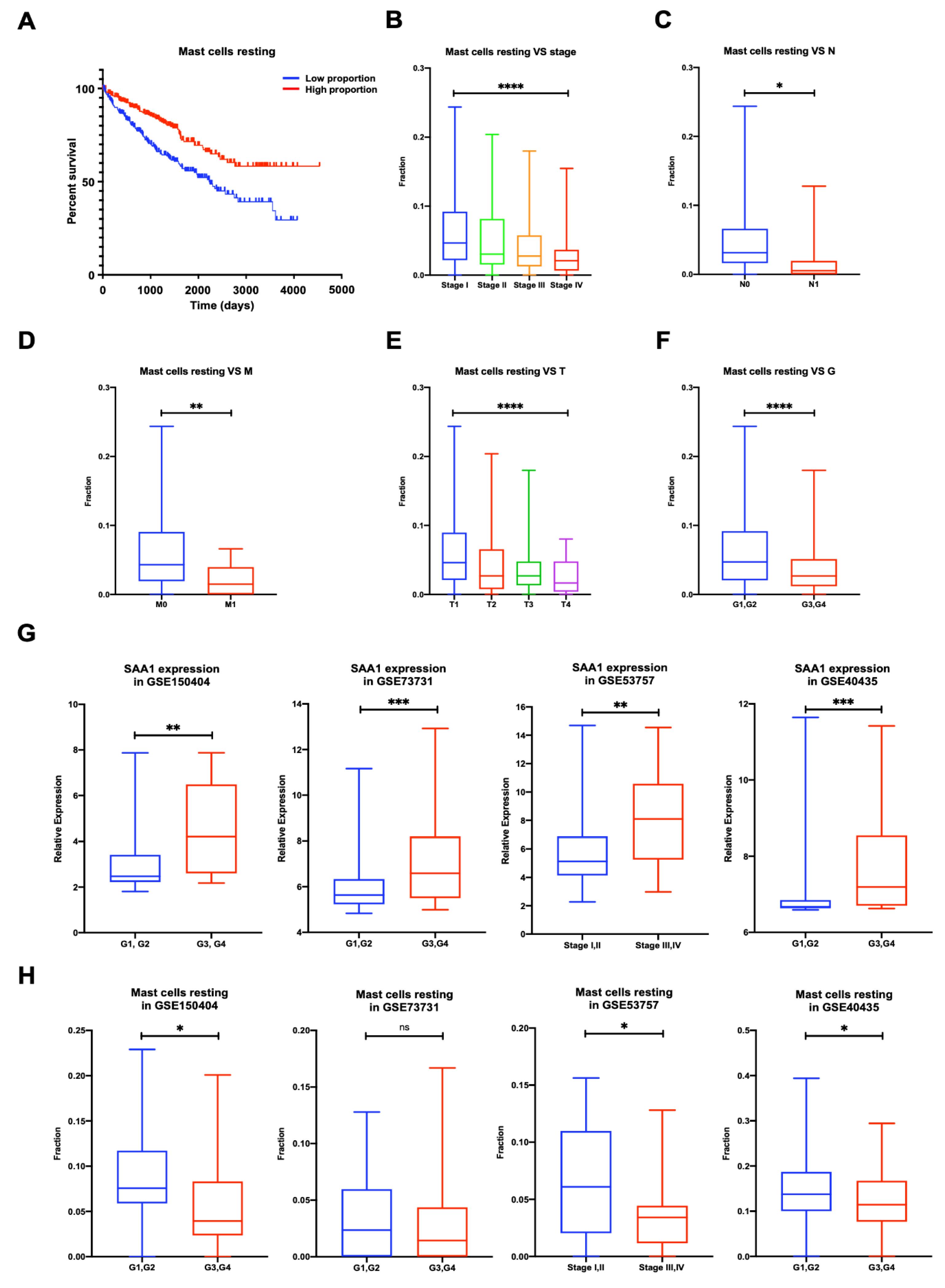
2.8. Proportion of Mast Cells Resting Was Significantly Associated with Favorable OS, Stage, and Metastases of ccRCC
2.9. Validation of the Correlation between SAA1 Expression, Mast Cells Resting Proportion, and Tumor Grade or Tumor Stage in Validation GEO Cohort
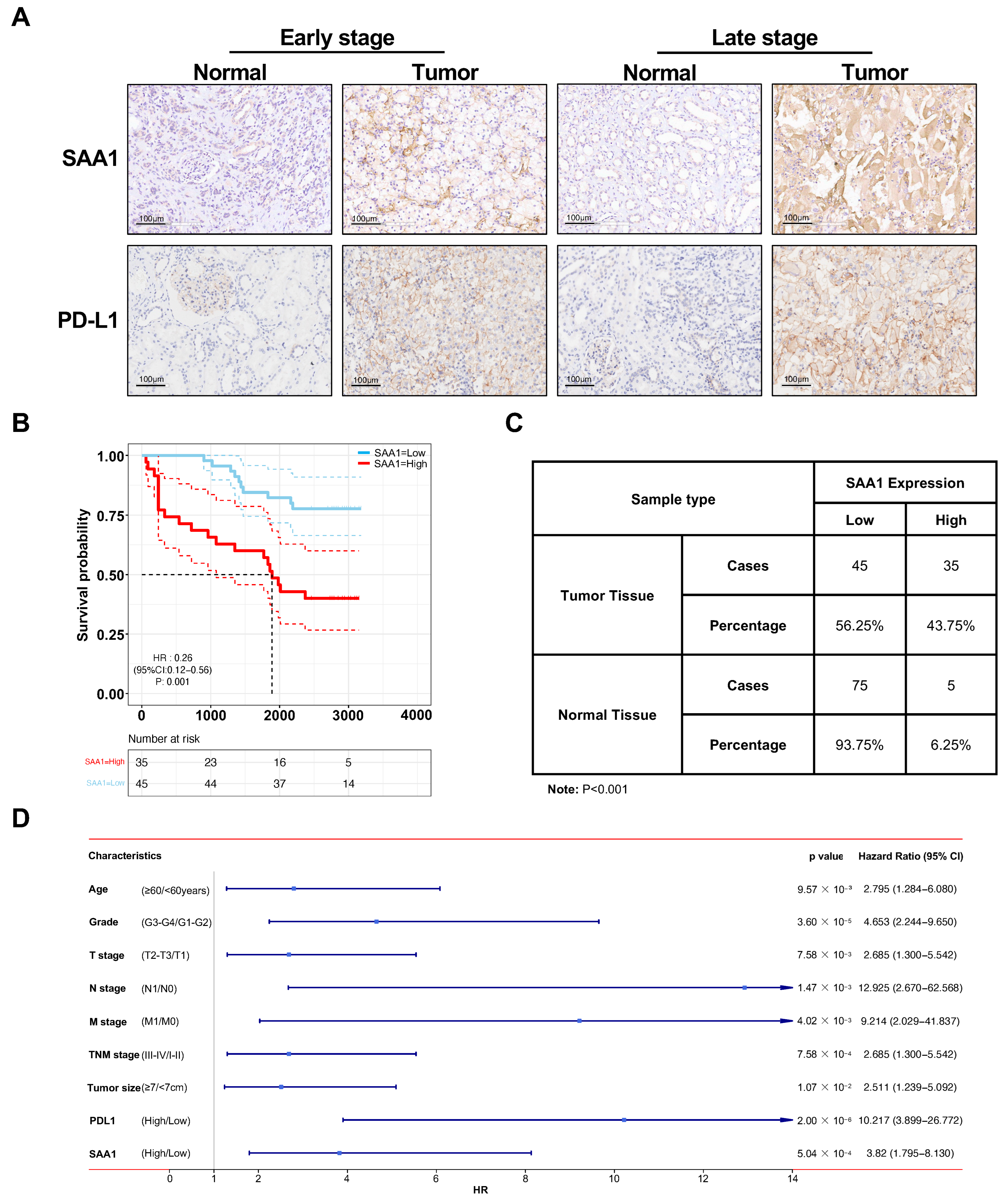
2.10. Validation of SAA1 Was Up-Regulated in ccRCC, Positively Correlated with PDL1 Expression, and Predicted Poor Therapeutic Effect
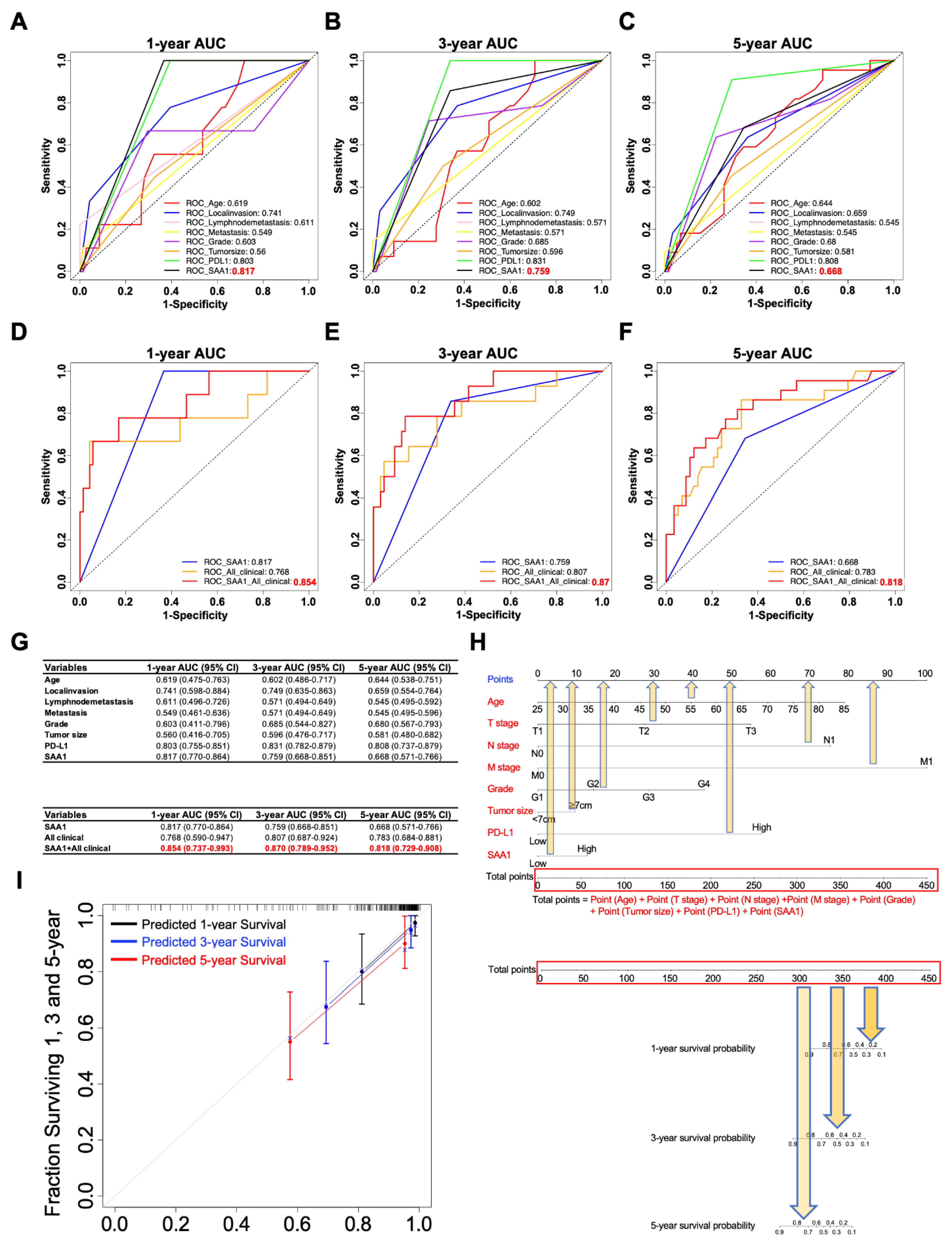
2.11. The Clinicopathologic Features in Combination with SAA1 Can Further Improve the Prediction Accuracy of Prognosis
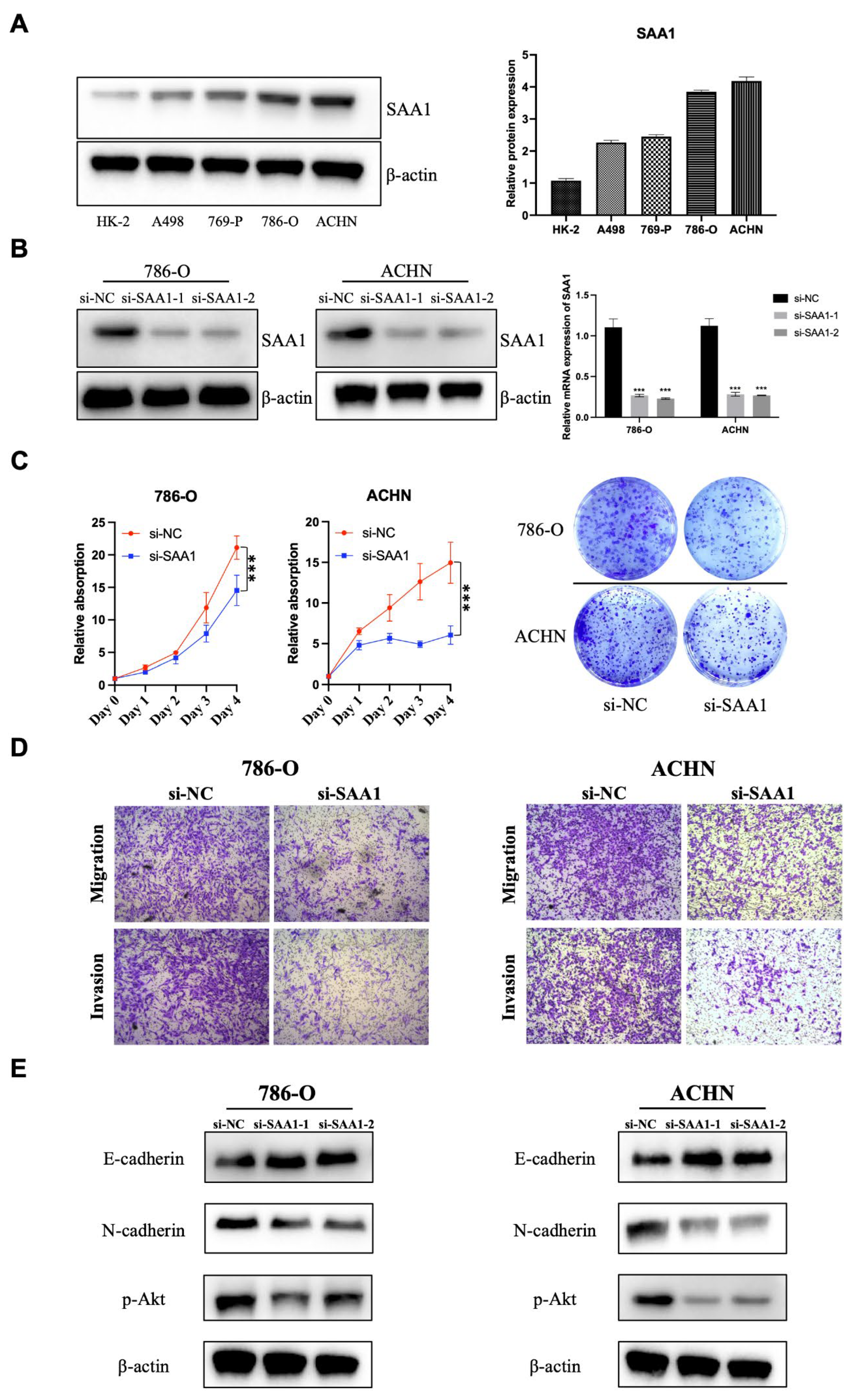
2.12. SAA1 Is Highly Expressed in ccRCC Cells and Promotes ccRCC Cell Proliferation, Migration, and Invasion
3. Discussion
4. Materials and Methods
4.1. Data Preparation
4.2. Calculation of ImmuneScore, StromalScore and ESTIMATEScore
4.3. Tumor-Infiltrating Immune Cells (TICs) Profile
4.4. Survival Analysis
4.5. Analysis of Differentially Expressed Genes (DEGs)
4.6. GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
4.7. Protein-Protein Interaction (PPI) Network Construction
4.8. Gene Set Enrichment Analysis
4.9. Tissue Samples and Immunohistochemistry
4.10. Cell Culture and Transfection
4.11. Cell Counting Kit-8 (CCK-8) and Colony Formation Assay
4.12. Transwell Migration and Invasion
4.13. Western Blot
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug. Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef]
- Wu, D.; Zhu, Z.Q.; Tang, H.X.; Shi, Z.E.; Kang, J.; Liu, Q.; Qi, J. Efficacy-shaping nanomedicine by loading Calcium Peroxide into Tumor Microenvironment-responsive Nanoparticles for the Antitumor Therapy of Prostate Cancer. Theranostics 2020, 10, 9808–9829. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Baine, M.K.; Turcu, G.; Zito, C.R.; Adeniran, A.J.; Camp, R.L.; Chen, L.; Kluger, H.M.; Jilaveanu, L.B. Characterization of tumor infiltrating lymphocytes in paired primary and metastatic renal cell carcinoma specimens. Oncotarget 2015, 6, 24990–25002. [Google Scholar] [CrossRef]
- Zhou, X.; Hou, W.; Gao, L.; Shui, L.; Yi, C.; Zhu, H. Synergies of Antiangiogenic Therapy and Immune Checkpoint Blockade in Renal Cell Carcinoma: From Theoretical Background to Clinical Reality. Front. Oncol. 2020, 10, 1321. [Google Scholar] [CrossRef] [PubMed]
- McGregor, B.A.; Lalani, A.A.; Xie, W.; Steinharter, J.A.; Bakuony, Z.E.; Martini, D.J.; Fleischer, J.H.; Abou-Alaiwi, S.; Nassar, A.; Nuzzo, P.V.; et al. Activity of cabozantinib after immune checkpoint blockade in metastatic clear-cell renal cell carcinoma. Eur. J. Cancer 2020, 135, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.H.; Li, K.W.; Chen, X.; He, H.X.; Peng, S.M.; Peng, S.R.; Wang, Q.; Li, Z.A.; Tao, Y.R.; Cai, W.L.; et al. HHLA2 and PD-L1 co-expression predicts poor prognosis in patients with clear cell renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Duan, Y.; Xia, M.; Dong, Y.; Chen, Y.; Zheng, L.; Chai, S.; Zhang, Q.; Wei, Z.; Liu, N.; et al. TFEB Mediates Immune Evasion and Resistance to mTOR Inhibition of Renal Cell Carcinoma via Induction of PD-L1. Clin. Cancer Res. 2019, 25, 6827–6838. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Wang, X.; Chai, H.; Wang, Z.; Lin, P.H.; Yao, Q.; Chen, C. Serum amyloid A induces endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2399–H2408. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Aino, M.; Kobayashi, S.I.; Okada, K.; Ohno, T.; Kikuchi, T.; Hayashi, J.I.; Yamamoto, G.; Hasegawa, Y.; Mitani, A. Serum Amyloid A Promotes E-Selectin Expression via Toll-Like Receptor 2 in Human Aortic Endothelial Cells. Mediat. Inflamm. 2016, 2016, 7150509. [Google Scholar] [CrossRef]
- De Buck, M.; Berghmans, N.; Portner, N.; Vanbrabant, L.; Cockx, M.; Struyf, S.; Opdenakker, G.; Proost, P.; Van Damme, J.; Gouwy, M. Serum amyloid A1alpha induces paracrine IL-8/CXCL8 via TLR2 and directly synergizes with this chemokine via CXCR2 and formyl peptide receptor 2 to recruit neutrophils. J. Leukoc. Biol. 2015, 98, 1049–1060. [Google Scholar] [CrossRef]
- Lv, M.; Xia, Y.F.; Li, B.; Liu, H.; Pan, J.Y.; Li, B.B.; Zhang, C.; An, F.S. Serum amyloid A stimulates vascular endothelial growth factor receptor 2 expression and angiogenesis. J. Physiol. Biochem. 2016, 72, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Izumi, Y.; Jiuchi, Y.; Kozuru, H.; Kawahara, C.; Nakamura, M.; Nakamura, T.; Agematsu, K.; Masumoto, J.; Yasunami, M.; et al. Serum amyloid A induces NLRP-3-mediated IL-1beta secretion in neutrophils. PLoS ONE 2014, 9, e96703. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.; Mullan, R.H.; McCormick, J.; Matthews, C.; Sullivan, O.; Kennedy, A.; FitzGerald, O.; Poole, A.R.; Bresnihan, B.; Veale, D.J.; et al. Acute-phase serum amyloid A regulates tumor necrosis factor alpha and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012, 64, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef]
- Gouwy, M.; De Buck, M.; Portner, N.; Opdenakker, G.; Proost, P.; Struyf, S.; Van Damme, J. Serum amyloid A chemoattracts immature dendritic cells and indirectly provokes monocyte chemotaxis by induction of cooperating CC and CXC chemokines. Eur. J. Immunol. 2015, 45, 101–112. [Google Scholar] [CrossRef]
- Xu, L.; Badolato, R.; Murphy, W.J.; Longo, D.L.; Anver, M.; Hale, S.; Oppenheim, J.J.; Wang, J.M. A novel biologic function of serum amyloid A. Induction of T lymphocyte migration and adhesion. J. Immunol. 1995, 155, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- McDermott, D.F.; Regan, M.M.; Clark, J.I.; Flaherty, L.E.; Weiss, G.R.; Logan, T.F.; Kirkwood, J.M.; Gordon, M.S.; Sosman, J.A.; Ernstoff, M.S.; et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2005, 23, 133–141. [Google Scholar] [CrossRef]
- Lai, Y.; Zhao, Z.; Zeng, T.; Liang, X.; Chen, D.; Duan, X.; Zeng, G.; Wu, W. Crosstalk between VEGFR and other receptor tyrosine kinases for TKI therapy of metastatic renal cell carcinoma. Cancer Cell. Int. 2018, 18, 31. [Google Scholar] [CrossRef]
- Baldewijns, M.M.; van Vlodrop, I.J.; Vermeulen, P.B.; Soetekouw, P.M.; van Engeland, M.; de Bruine, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Leite, K.R.; Reis, S.T.; Junior, J.P.; Zerati, M.; Gomes Dde, O.; Camara-Lopes, L.H.; Srougi, M. PD-L1 expression in renal cell carcinoma clear cell type is related to unfavorable prognosis. Diagn. Pathol. 2015, 10, 189. [Google Scholar] [CrossRef]
- Thompson, R.H.; Dong, H.; Lohse, C.M.; Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin. Cancer Res. 2007, 13, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug. Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Knebel, F.H.; Uno, M.; Galatro, T.F.; Belle, L.P.; Oba-Shinjo, S.M.; Marie, S.K.N.; Campa, A. Serum amyloid A1 is upregulated in human glioblastoma. J. Neurooncol 2017, 132, 383–391. [Google Scholar] [CrossRef]
- Lin, C.Y.; Yang, S.T.; Shen, S.C.; Hsieh, Y.C.; Hsu, F.T.; Chen, C.Y.; Chiang, Y.H.; Chuang, J.Y.; Chen, K.Y.; Hsu, T.I.; et al. Serum amyloid A1 in combination with integrin alphaVbeta3 increases glioblastoma cells mobility and progression. Mol. Oncol. 2018, 12, 756–771. [Google Scholar] [CrossRef]
- Walter, M.; Szendrői, A.; Kroeger, N.; Bode, C.; Steiner, T.; Kniemeyer, O.; Zipfel, P.; Romics, I.; Grimm, M.-O.; Junker, K. 315 Prediction of TKI-therapy response in patients with metastatic renal cell carcinoma by serum proteomics. J. Urol. 2012, 187, e722. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, H.; Cheng, N.; Qian, F.; Ye, R.D. Serum amyloid A1 isoforms display different efficacy at Toll-like receptor 2 and formyl peptide receptor 2. Immunobiology 2014, 219, 916–923. [Google Scholar] [CrossRef]
- Facci, L.; Barbierato, M.; Zusso, M.; Skaper, S.D.; Giusti, P. Serum amyloid A primes microglia for ATP-dependent interleukin-1beta release. J. Neuroinflamm. 2018, 15, 164. [Google Scholar] [CrossRef]
- Li, Y.; Cai, L.; Wang, H.; Wu, P.; Gu, W.; Chen, Y.; Hao, H.; Tang, K.; Yi, P.; Liu, M.; et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011, 30, 3887–3899. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, H.; Zhu, Z.; Yan, Q.; Wang, L.; Liang, Q.; Ye, R.D. Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J. Immunol. 2015, 194, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Poeta, M.L.; Costantini, M.; Pfeiffer, R.M.; Li, M.; Golubeva, Y.; Lawrence, S.; Mutreja, K.; Amoreo, C.; Dabrowska, M.; et al. Characterizing the tumor microenvironment in rare renal cancer histological types. J. Pathol. Clin. Res. 2022, 8, 88–98. [Google Scholar] [CrossRef]
- Zheng, B.; Xie, F.; Cheng, F.; Wang, J.; Yao, Z.; He, W.; Niu, Z. Integrative Analysis of Immune-Related Genes in the Tumor Microenvironment of Renal Clear Cell Carcinoma and Renal Papillary Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 760031. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | No. of Patients | SAA1 Expression | p Value a | |
|---|---|---|---|---|
| Low | High | |||
| All patients | 80 | 45 | 35 | |
| Age (years) | 0.499 | |||
| <60 | 40 | 21 | 19 | |
| ≥60 | 40 | 24 | 16 | |
| Gender | 0.194 | |||
| Male | 52 | 32 | 20 | |
| Female | 28 | 13 | 15 | |
| Grade | 0.017 * | |||
| 1 | 20 | 9 | 11 | |
| 2 | 33 | 25 | 8 | |
| 3 | 26 | 10 | 16 | |
| 4 | 1 | 1 | 0 | |
| T classification | 0.038 * | |||
| T1 | 45 | 30 | 15 | |
| T2 | 29 | 14 | 15 | |
| T3 | 6 | 1 | 5 | |
| Lymphatic metastasis (N) | 0.188 | |||
| N0 | 78 | 45 | 33 | |
| N1 | 2 | 0 | 2 | |
| Distant metastasis (M) | 0.188 | |||
| M0 | 78 | 45 | 33 | |
| M1 | 2 | 0 | 2 | |
| AJCC stage | 0.035 * | |||
| I | 45 | 30 | 15 | |
| II | 27 | 14 | 13 | |
| III | 6 | 1 | 5 | |
| IV | 2 | 0 | 2 | |
| Tumor size | 0.046 * | |||
| <7 cm | 53 | 34 | 19 | |
| ≥7 cm | 27 | 11 | 16 | |
| PDL1 expression | <0.001 *** | |||
| Low | 43 | 32 | 11 | |
| High | 37 | 13 | 24 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Wu, Y.; Fu, G.; Chen, X.; Sun, J.; Tian, J.; Jiang, P.; Wang, Y.; Jin, B. SAA1 Has Potential as a Prognostic Biomarker Correlated with Cell Proliferation, Migration, and an Indicator for Immune Infiltration of Tumor Microenvironment in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 7505. https://doi.org/10.3390/ijms24087505
Xu Z, Wu Y, Fu G, Chen X, Sun J, Tian J, Jiang P, Wang Y, Jin B. SAA1 Has Potential as a Prognostic Biomarker Correlated with Cell Proliferation, Migration, and an Indicator for Immune Infiltration of Tumor Microenvironment in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(8):7505. https://doi.org/10.3390/ijms24087505
Chicago/Turabian StyleXu, Zhijie, Yunfei Wu, Guanghou Fu, Xiaoyi Chen, Junjie Sun, Junjie Tian, Peng Jiang, Yimin Wang, and Baiye Jin. 2023. "SAA1 Has Potential as a Prognostic Biomarker Correlated with Cell Proliferation, Migration, and an Indicator for Immune Infiltration of Tumor Microenvironment in Clear Cell Renal Cell Carcinoma" International Journal of Molecular Sciences 24, no. 8: 7505. https://doi.org/10.3390/ijms24087505
APA StyleXu, Z., Wu, Y., Fu, G., Chen, X., Sun, J., Tian, J., Jiang, P., Wang, Y., & Jin, B. (2023). SAA1 Has Potential as a Prognostic Biomarker Correlated with Cell Proliferation, Migration, and an Indicator for Immune Infiltration of Tumor Microenvironment in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences, 24(8), 7505. https://doi.org/10.3390/ijms24087505