
Comparative Transcriptome Analysis Reveals the Effect of the DHN Melanin Biosynthesis Pathway on the Appressorium Turgor Pressure of the Poplar Anthracnose-Causing Fungus Colletotrichum gloeosporioides

Abstract
1. Introduction
2. Results
2.1. Appressorium Morphology in Different Developmental Stages
2.2. Changes in the Appressorium Turgor Pressure in the Wild-Type Control and Mutants and CgCmr1 and CgPks1 Expression in the Wild-Type Control at Different Time Points
2.3. Transcriptome Sequencing and Data Assembly
2.4. Differentially Expressed Genes among the Transcriptomes
2.5. GO Enrichment Analysis of the DEGs
2.6. Enriched KEGG Pathways among the DEGs
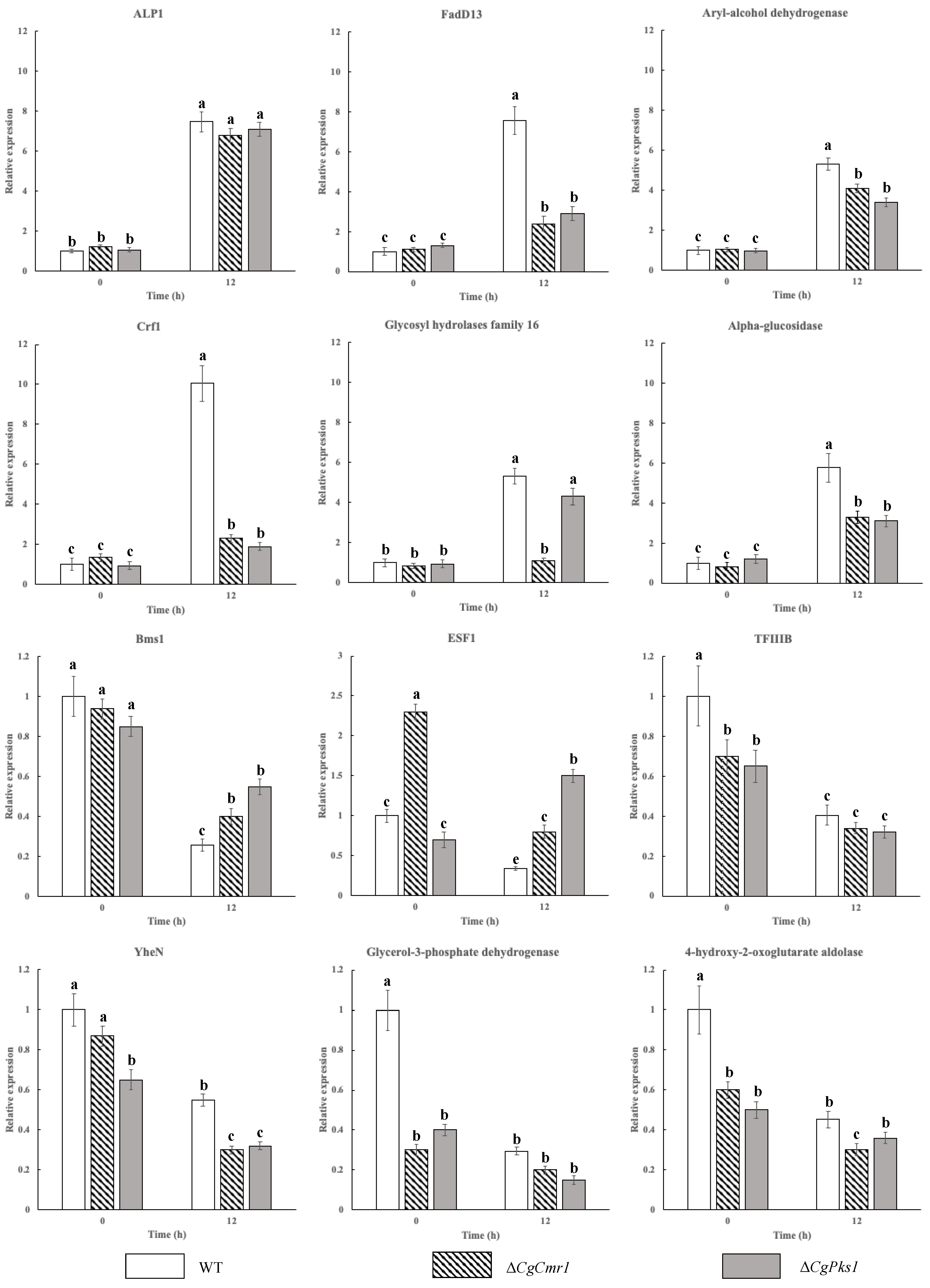
2.7. Verification of Gene Expression by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Determination of the Turgor Pressure in WT, ΔCgCmr1, and ΔCgPks1 Appressoria at Different Time Points
4.3. Total RNA Isolation and cDNA Synthesis
4.4. Sequencing and Assembly of the C. gloeosporioides Transcriptome
4.5. Identification of Enriched GO Terms and KEGG Pathways
4.6. Quantitative Real-Time PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Sun, H.; Pei, J.; Dong, Y.; Wang, F.; Chen, H.; Sun, Y.; Wang, N.; Li, H.; Li, Y. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene 2012, 511, 54–61. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Z.; Wu, J.; Ji, Z.; Zhang, J. Comparative transcriptome analysis reveals significant differences in gene expression between appressoria and hyphae in Colletotrichum gloeosporioides. Gene 2018, 670, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ji, Z.; Wang, N.; Chi, F.; Xu, C.; Zhou, Z.; Zhang, J. Identification of conidiogenesis-associated genes in Colletotrichum gloeosporioides by Agrobacterium tumefaciens-mediated transformation. Curr. Microbiol. 2016, 73, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Barhoom, S.; Sharon, A. cAMP regulation of “pathogenic” and “saprophytic” fungal spore germination. Fungal Genet. Biol. 2004, 41, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.S.; Abreu, M.S.; Alves, E.; Ferreira, J.B. Histopathological studies of the interaction Colletotrichum gloeosporioides: Coffee tree. Bragantia 2009, 68, 117–123. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; He, H.; Wang, X.; Tian, C. Histopathology study of poplar leaves infected by Colletotrichum gloeosporioides. J. Beijing For. Univ. 2018, 40, 101–109. [Google Scholar]
- Lin, F.C. Cell biology of appressorium formation of Magnapothe grisea. Acta Phytopathol. Sin. 2001, 2, 97–101. [Google Scholar]
- Wang, Z.Y.; Jenkinson, J.M.; Holcombe, L.J.; Soanes, D.M.; Veneault-Fourrey, C.; Bhambra, G.K.; Talbot, N.J. The molecular biology of appressorium turgor generation by the rice blast fungus Magnaporthe grisea. Biochem. Soc. Trans. 2005, 33, 384–388. [Google Scholar] [CrossRef]
- Ryder, L.S.; Talbot, N.J. Regulation of appressorium development in pathogenic fungi. Curr. Opin. Plant Biol. 2015, 26, 8–13. [Google Scholar] [CrossRef]
- He, P.H.Z.; Wang, Y.L.; Wang, X.L.; Zhang, X.; Tian, C. The mitogen-activated protein kinase CgMK1 governs appressorium formation, melanin synthesis, and plant infection of Colletotrichum gloeosporioides. Front. Microbiol. 2017, 8, 2216. [Google Scholar] [CrossRef]
- Wang, K.; Wang, W.; Zheng, F. Advance research on appressorium of Colletotrichum. Chin. Agric. Sci. Bull. 2007, 1, 265–270. [Google Scholar]
- Fang, Y.L.; Xia, L.M.; Wang, P.; Zhu, L.-H.; Ye, J.-R.; Huang, L. The MAPKKK CgMck1 is required for cell wall integrity, appressorium development, and pathogenicity in Colletotrichum gloeosporioides. Genes 2018, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, B.; Pan, Y.T.; Zhang, Y.Z.; Li, D.W.; Huang, L. Calcineurin-responsive transcription factor CgCrzA is required for cell wall integrity and infection-related morphogenesis in Colletotrichum gloeosporioides. Plant Pathol. J. 2020, 36, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Meng, F.L.; Tian, C.M. Transcriptional network in Colletotrichum gloeosporioides mutants lacking Msb2 or Msb2 and Sho1. J. Fungi 2022, 8, 207. [Google Scholar] [CrossRef]
- Li, Y.F.; He, P.H.Z.; Tian, C.M.; Wang, Y. CgHog1 controls the adaptation to both sorbitol and fludioxonil in Colletotrichum gloeosporioides. Fungal Genet. Biol. 2020, 135, 103289. [Google Scholar] [CrossRef]
- Wang, X.L.; Lu, D.X.; Tian, C.M. Analysis of melanin biosynthesis in the plant pathogenic fungus Colletotrichum gloeosporioides. Fungal Biol. 2021, 125, 679–692. [Google Scholar] [CrossRef]
- Henson, J.M.; Butler, M.J.; Day, A.W. The dark side of the mycelium: Melanins of phytopathogenic fungi. Annu. Rev. Phytopathol. 1999, 37, 447–471. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, B.; Hua, C.; Meng, P.; Wang, S.; Chen, Z.; Du, Y.; Gao, F.; Huang, J. VdPKS1 is required for melanin formation and virulence in a cotton wilt pathogen Verticillium dahliae. Sci. China Life Sci. 2017, 60, 868–879. [Google Scholar] [CrossRef]
- Schumacher, J. DHN melanin biosynthesis in the plant pathogenic fungus Botrytis cinerea is based on two developmentally regulated key enzyme (PKS)-encoding genes. Mol. Microbiol. 2016, 99, 729–748. [Google Scholar] [CrossRef]
- Scindiya, M.; Malathi, P.; Kaverinathan, K.; Sundar, A.R.; Viswanathan, R. RNA-mediated silencing of PKS1 gene in Colletotrichum falcatum causing red rot in sugarcane. Eur. J. Plant Pathol. 2019, 153, 371–384. [Google Scholar] [CrossRef]
- Takano, Y.; Kubo, Y.; Shimizu, K.; Mise, K.; Okuno, T.; Furusawa, I. Structural analysis of PKS1, a polyketide synthase gene involved in melanin biosynthesis in Colletotrichum lagenarium. Mol. Gen. Genet. 1995, 249, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Kenmochi, Y.; Takano, Y.; Sweigard, J.; Farrall, L.; Furusawa, I.; Horino, O.; Kubo, Y. Novel fungal transcriptional activators, Cmr1p of Colletotrichum lagenarium and Pig1p of Magnaporthe grisea, contain Cys2His2 zinc finger and Zn (II) 2Cys6 binuclear cluster DNA-binding motifs and regulate transcription of melanin biosynthesis genes in a developmentally specific manner. Mol. Microbiol. 2000, 38, 940–954. [Google Scholar]
- Wang, Y.; Hu, X.; Fang, Y.; Anchieta, A.; Goldman, P.H.; Hernandez, G.; Klosterman, S.J. Transcription factor VdCmr1 is required for pigment production, protection from UV irradiation, and regulates expression of melanin biosynthetic genes in Verticillium dahliae. Microbiology 2018, 164, 685. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Fukada, J.; Moriwaki, A.; Fujikawa, T.; Ohashi, M.; Hibi, T.; Hayashi, N. Mstu1, an APSES transcription factor, is required for appressorium-mediated infection in Magnaporthe grisea. Biosci. Biotechnol. Biochem. 2009, 73, 1779–1786. [Google Scholar] [CrossRef]
- Cao, H.J.; Huang, P.Y.; Yan, Y.X.; Shi, Y.; Dong, B.; Liu, X.; Ye, L.; Lin, F.; Lu, J. The basic helix-loop-helix transcription factor Crf1 is required for development and pathogenicity of the rice blast fungus by regulating carbohydrate and lipid metabolism. Environ. Microbiol. 2018, 20, 3427–3441. [Google Scholar] [CrossRef] [PubMed]
- Thines, E.; Weber, R.W.; Talbot, N.J. MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell 2000, 12, 1703–1718. [Google Scholar]
- Wilson, R.; Talbot, N. Under pressure: Investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol. 2009, 7, 185–195. [Google Scholar] [CrossRef]
- Liu, X.H.; Liang, S.; Wei, Y.Y.; Zhu, X.M.; Li, L.; Liu, P.P.; Zheng, Q.X.; Zhou, H.N.; Zhang, Y.; Mao, L.J.; et al. Metabolomics analysis identifies sphingolipids as key signaling moieties in appressorium morphogenesis and function in Magnaporthe oryzae. mBio 2019, 10, e01467-19. [Google Scholar] [CrossRef]
- Ferreira, R.B.; Monteiro, S.; Freitas, R.; Santos, C.N.; Chen, Z.; Batista, L.M.; Duarte, J.; Borges, A.; Teixeira, A.R. Fungal pathogens: The battle for plant infection. Crit. Rev. Plant Sci. 2006, 25, 505–524. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef]
- Su, Y.L.; Li, J.M.; Li, M.; Luan, J.B.; Ye, X.D.; Wang, X.W.; Liu, S.S. Transcriptomic analysis of the salivary glands of an invasive whitefly. PLoS ONE 2012, 7, e39303. [Google Scholar] [CrossRef]
- Yin, Y.; Martin, J.; Abubucker, S.; Scott, A.L.; McCarter, J.P.; Wilson, R.K.; Jasmer, D.P.; Mitreva, M. Intestinal transcriptomes of nematodes: Comparison of the parasites Ascaris suum and Haemonchus contortus with the free-living Caenorhabditis elegans. PLoS Negl. Trop. Dis. 2008, 2, e269. [Google Scholar] [CrossRef]
- Li, Z.; Liang, Y.M.; Tian, C.M. Characterization of the causal agent of poplar anthracnose occurring in the Beijing region. Mycotax 2012, 120, 277–286. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Data (bp) | Clean Reads | Q30 (%) | Total Mapping (%) | Uniquely Mapping (%) |
|---|---|---|---|---|---|
| CgWT0h | 7,534,480,292 | 25,188,447 | 94.46 | 91.76 | 91.38 |
| CgWT12h | 6,803,953,388 | 22,770,082 | 94.81 | 93.11 | 92.85 |
| Condition | ID | log2FC | p-Value | Annoation |
|---|---|---|---|---|
| CgWT0h-CgWT12h-up | EVM0007582 | 6.3931 | 8.68 × 10−7 | Fum1, highly reducing polyketide synthase |
| EVM0002071 | 5.3559 | 8.20 × 10−5 | Acyl-CoA-binding protein | |
| EVM0007891 | 5.2385 | 9.51 × 10−5 | AtmM, FAD-dependent monooxygenase | |
| EVM0001628 | 5.1897 | 1.13 × 10−4 | Asl1, alkali-sensitive linkage protein | |
| EVM0011845 | 4.8421 | 2.75 × 10−4 | FadD13, long-chain-fatty-acid-CoA ligase | |
| EVM0001548 | 4.4904 | 9.39 × 10−4 | ApdG, acyl-CoA dehydrogenase | |
| EVM0011186 | 4.4468 | 7.35 × 10−4 | Nrk1, nicotinamide riboside kinase | |
| EVM0005520 | 4.3341 | 8.93 × 10−4 | Fkr-3, FK506-binding protein | |
| EVM0001282 | 4.1999 | 1.41 × 10−3 | 4-hydroxy-2-oxoglutarate aldolase | |
| EVM0006481 | 4.0481 | 2.77 × 10−3 | Multifunctional cytochrome P450 monooxygenase | |
| CgWT0h-CgWT12h-down | EVM0008257 | 5.6860 | 2.04 × 10−5 | PelA, pectate trisaccharide-lyase |
| EVM0008999 | 5.0929 | 1.84 × 10−4 | Presilphiperfolan-8-beta-ol synthase | |
| EVM0001567 | 4.5656 | 1.06 × 10−3 | DIT1, spore wall maturation protein | |
| EVM0009287 | 4.4408 | 1.40 × 10−3 | Nmral1, NmrA-like family domain-containing protein | |
| EVM0007786 | 4.4165 | 1.33 × 10−3 | Sodium transport ATPase 5 | |
| EVM0005978 | 4.3315 | 1.66 × 10−3 | Xyn1, endo-1,4-beta-xylanase | |
| EVM0006498 | 4.2992 | 1.67 × 10−3 | SDH-4, L-sorbose-1-dehydrogenase | |
| EVM0013274 | 4.2895 | 1.75 × 10−3 | Transporter | |
| EVM0001800 | 3.8492 | 5.65 × 10−3 | Sulfoquinovose-1-dehydrogenase | |
| EVM0011157 | 3.8457 | 5.28 × 10−3 | Multicopper oxidase |
| KEGG (KO) Term | No. Genes |
|---|---|
| Carbon metabolism | 201 |
| Biosynthesis of amino acids | 198 |
| Tryptophan metabolism | 153 |
| Glycine, serine and threonine metabolism | 138 |
| Fatty acid metabolism | 130 |
| Phenylpropanoid biosynthesis | 126 |
| Tyrosine metabolism | 121 |
| Arginine and proline metabolism | 111 |
| Glycerophospholipid metabolism | 99 |
| Carotenoid biosynthesis | 96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, X.; Tian, C.; Meng, F. Comparative Transcriptome Analysis Reveals the Effect of the DHN Melanin Biosynthesis Pathway on the Appressorium Turgor Pressure of the Poplar Anthracnose-Causing Fungus Colletotrichum gloeosporioides. Int. J. Mol. Sci. 2023, 24, 7411. https://doi.org/10.3390/ijms24087411
Qin X, Tian C, Meng F. Comparative Transcriptome Analysis Reveals the Effect of the DHN Melanin Biosynthesis Pathway on the Appressorium Turgor Pressure of the Poplar Anthracnose-Causing Fungus Colletotrichum gloeosporioides. International Journal of Molecular Sciences. 2023; 24(8):7411. https://doi.org/10.3390/ijms24087411
Chicago/Turabian StyleQin, Xinyu, Chengming Tian, and Fanli Meng. 2023. "Comparative Transcriptome Analysis Reveals the Effect of the DHN Melanin Biosynthesis Pathway on the Appressorium Turgor Pressure of the Poplar Anthracnose-Causing Fungus Colletotrichum gloeosporioides" International Journal of Molecular Sciences 24, no. 8: 7411. https://doi.org/10.3390/ijms24087411
APA StyleQin, X., Tian, C., & Meng, F. (2023). Comparative Transcriptome Analysis Reveals the Effect of the DHN Melanin Biosynthesis Pathway on the Appressorium Turgor Pressure of the Poplar Anthracnose-Causing Fungus Colletotrichum gloeosporioides. International Journal of Molecular Sciences, 24(8), 7411. https://doi.org/10.3390/ijms24087411