

SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
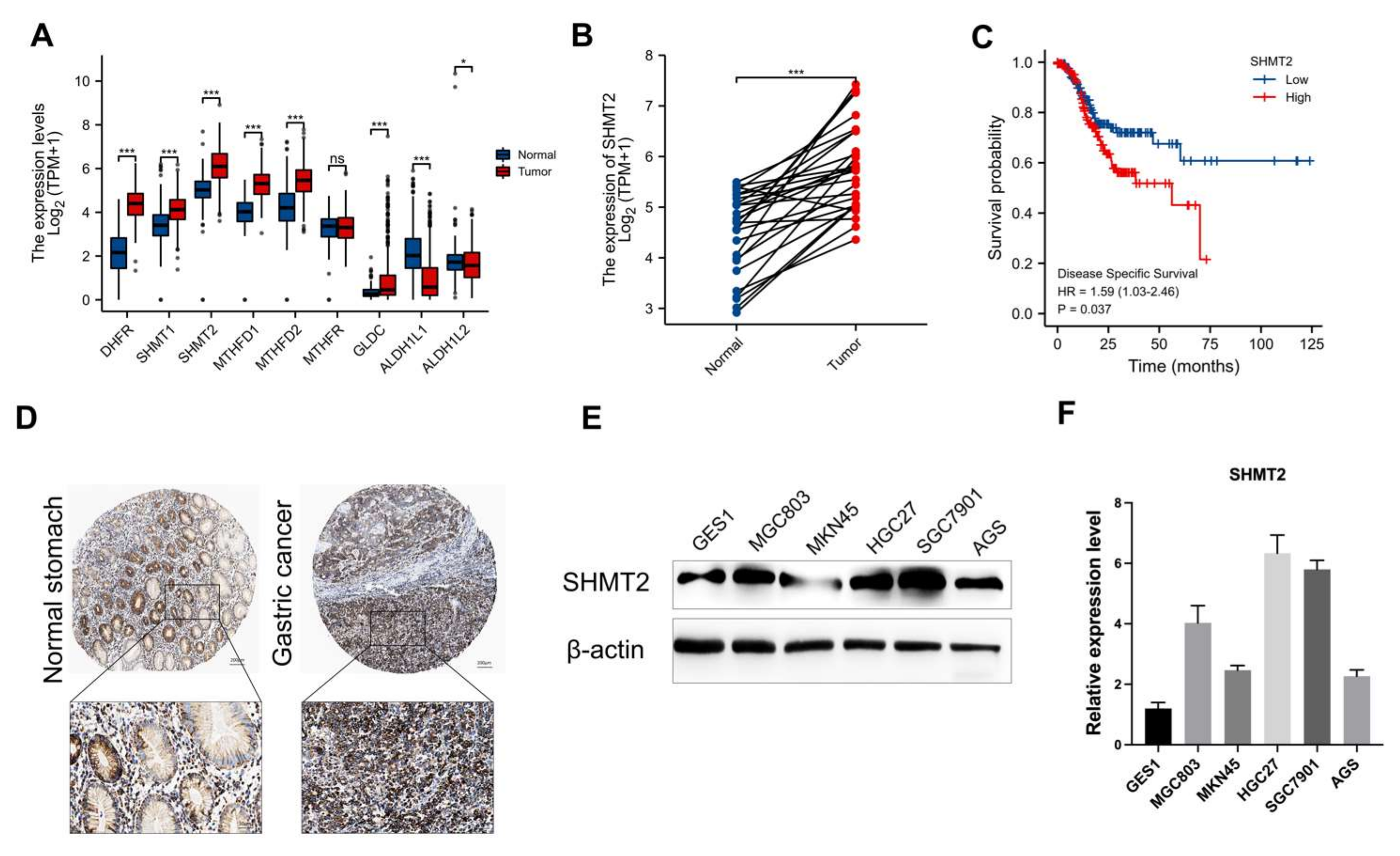
2.1. SHMT2 Was Highly Expressed in Gastric Cancer Patients and Was Accompanied by a Poor Prognosis
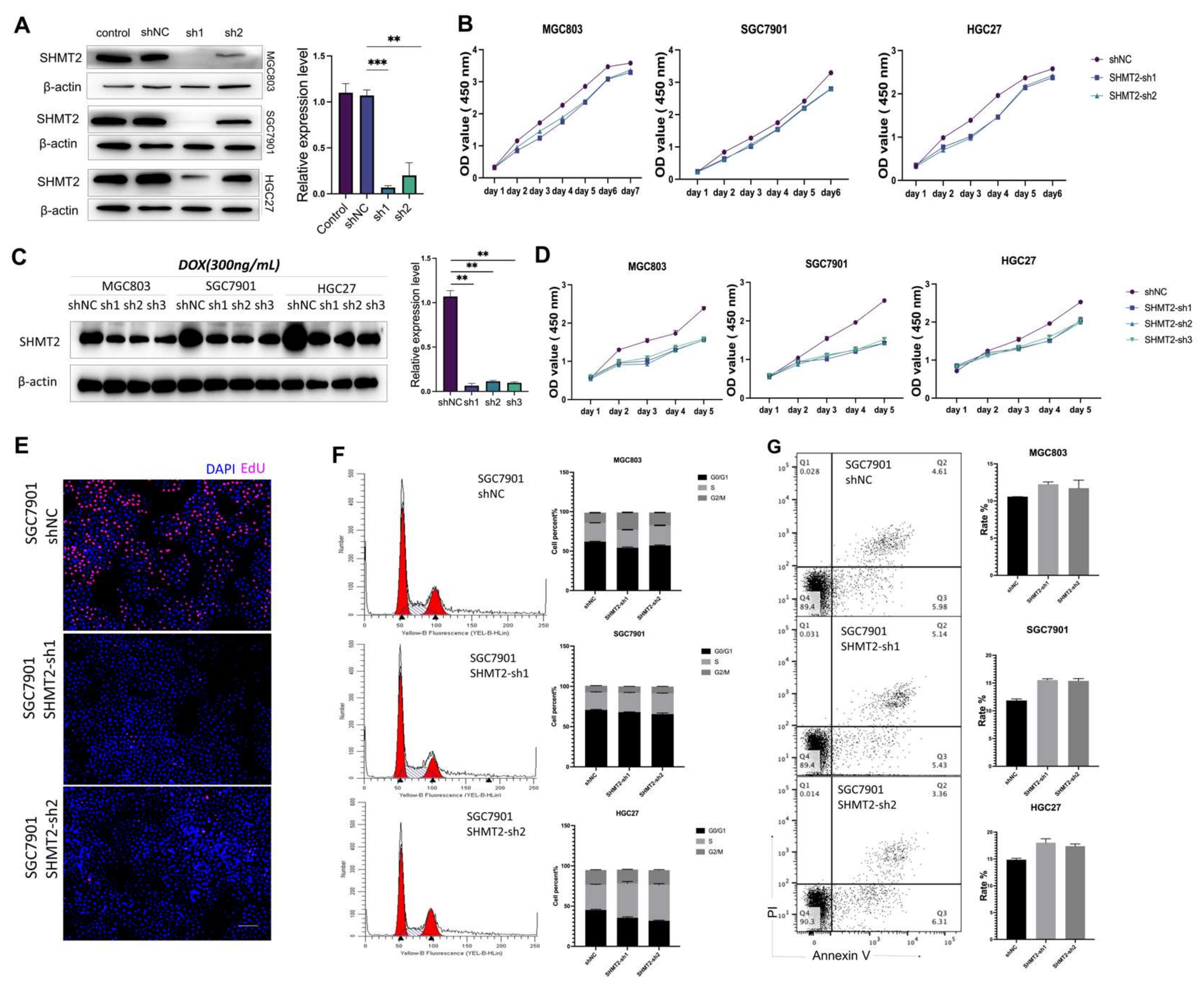
2.2. The Proliferation of Gastric Cancer Cells In Vitro Was Impacted by the Deletion of SHMT2
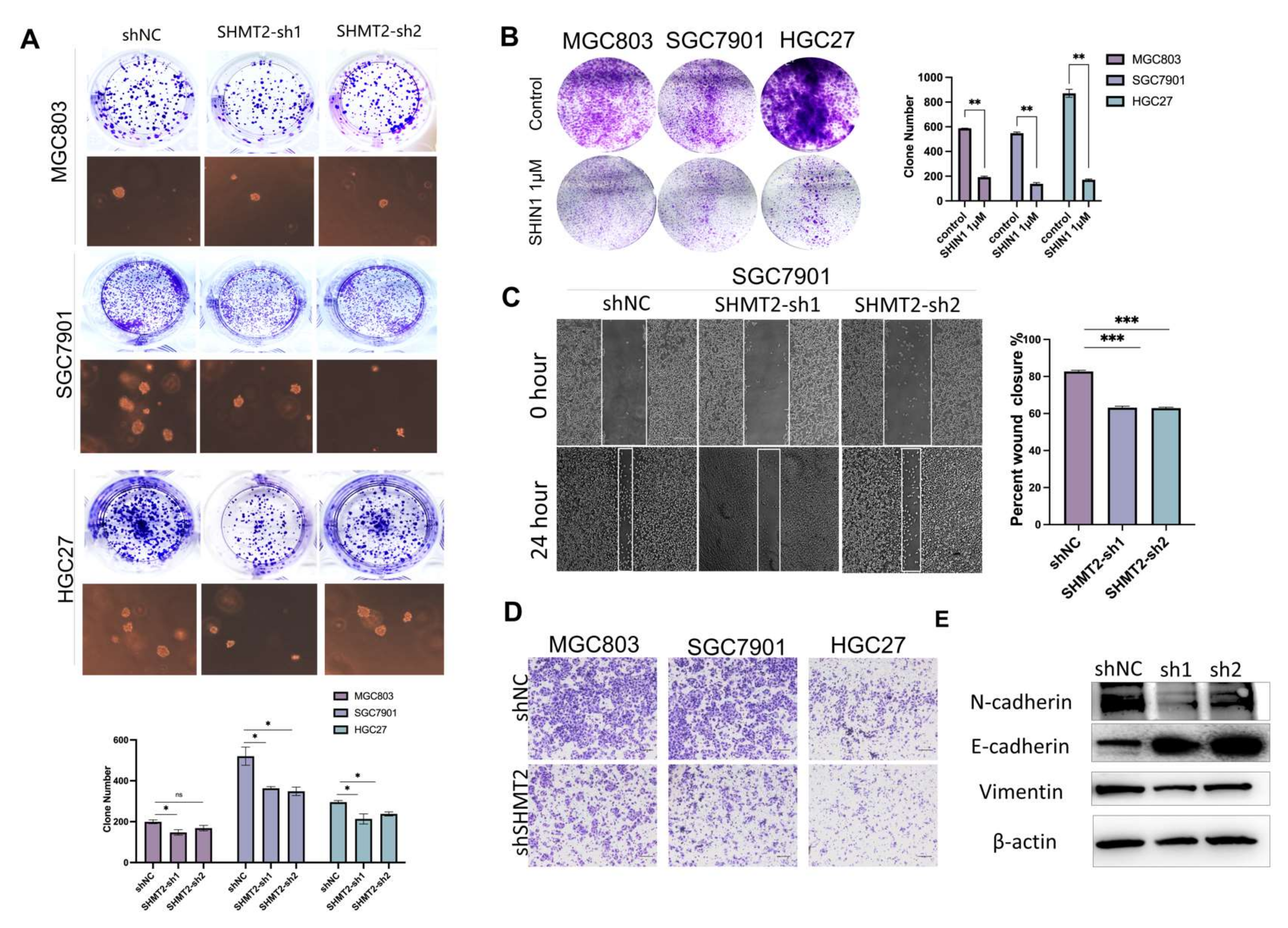
2.3. SHMT2 Deletion Affected the Ability of Gastric Cancer Cells to Invade and Metastasize
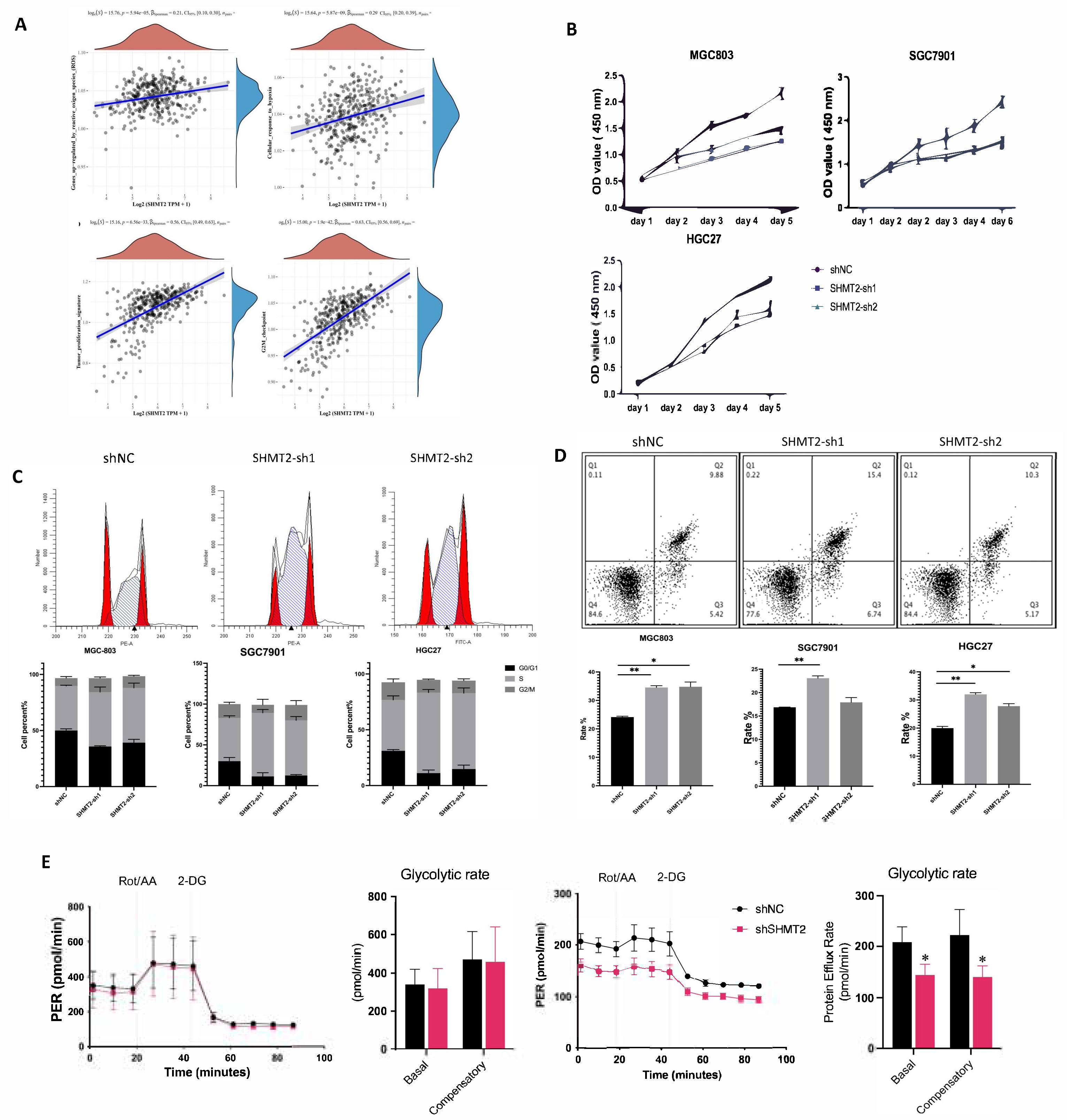
2.4. SHMT2 Deficiency Affected Cell Proliferation, Cell Cycle, Apoptosis, Invasion, and Metastasis, Together with a Major Impact on Glycolysis in Hypoxic Environments
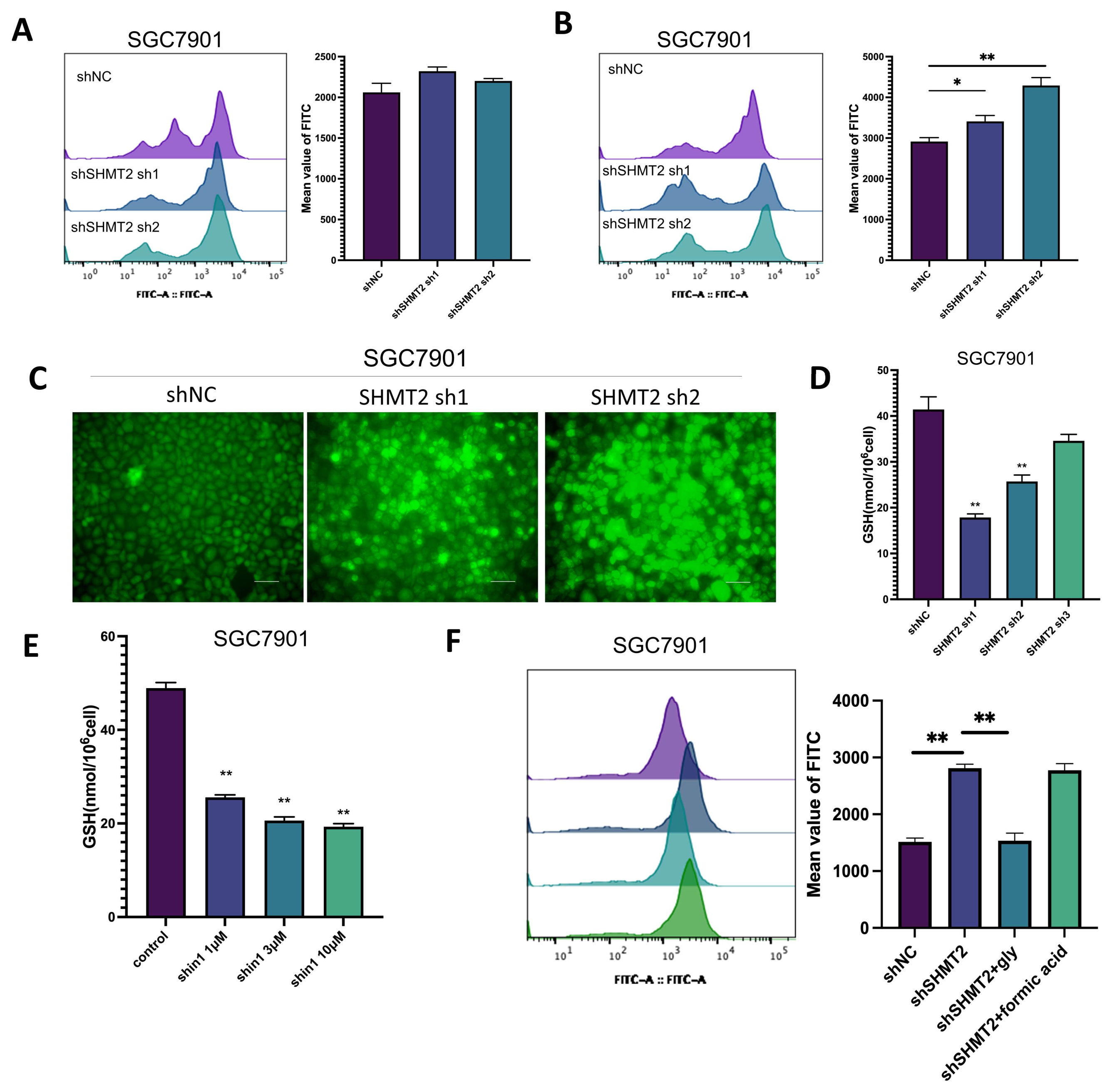
2.5. SHMT2 Maintained Cell Growth by Sustaining an Oxidative Reduction in Gastric Cancer Cells under Hypoxia
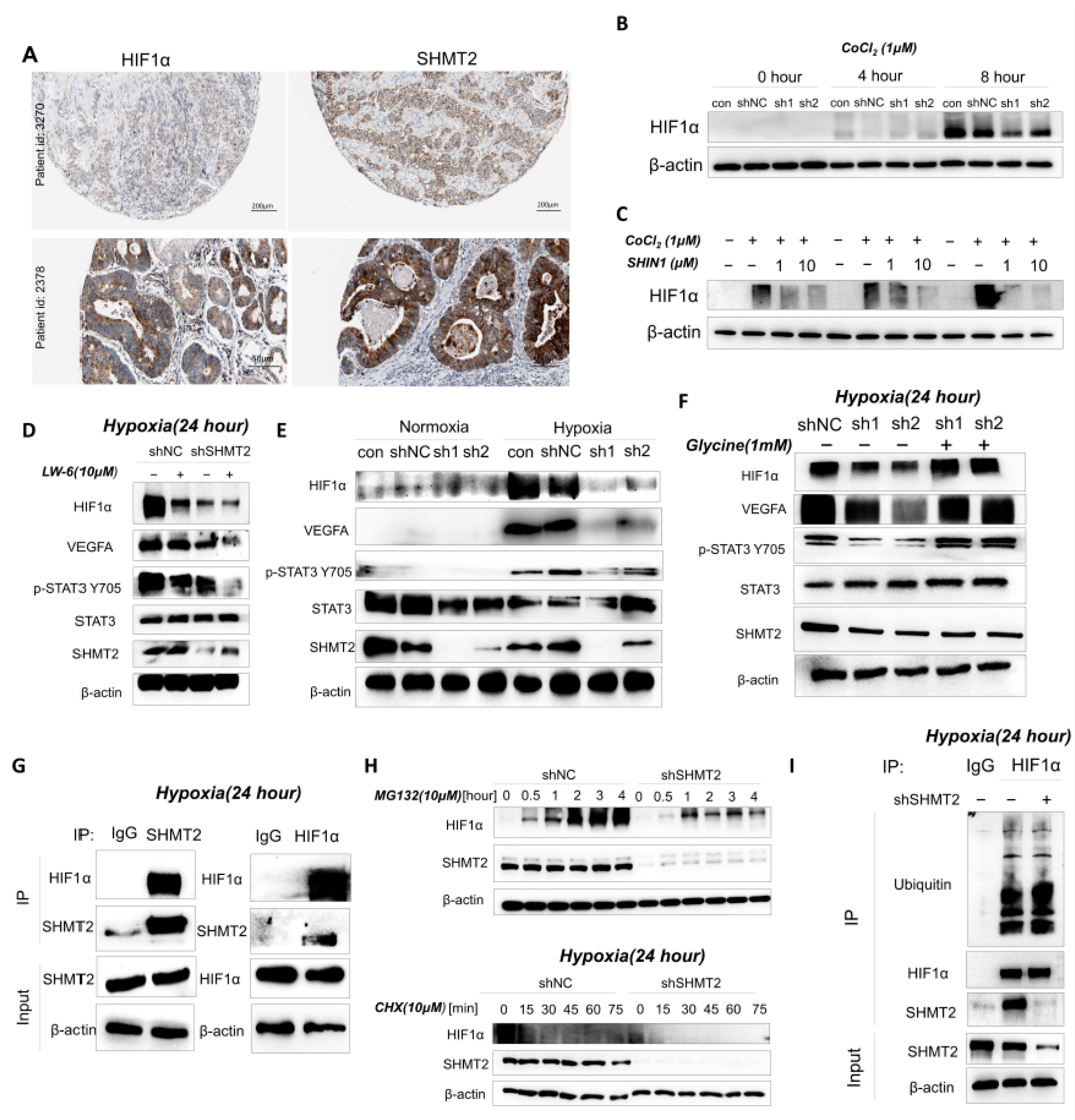
2.6. SHMT2 Contributed to Cell Proliferation under Hypoxia by Maintaining the Redox Balance of Cells via the HIF1a/VEGF/STAT3 Pathway
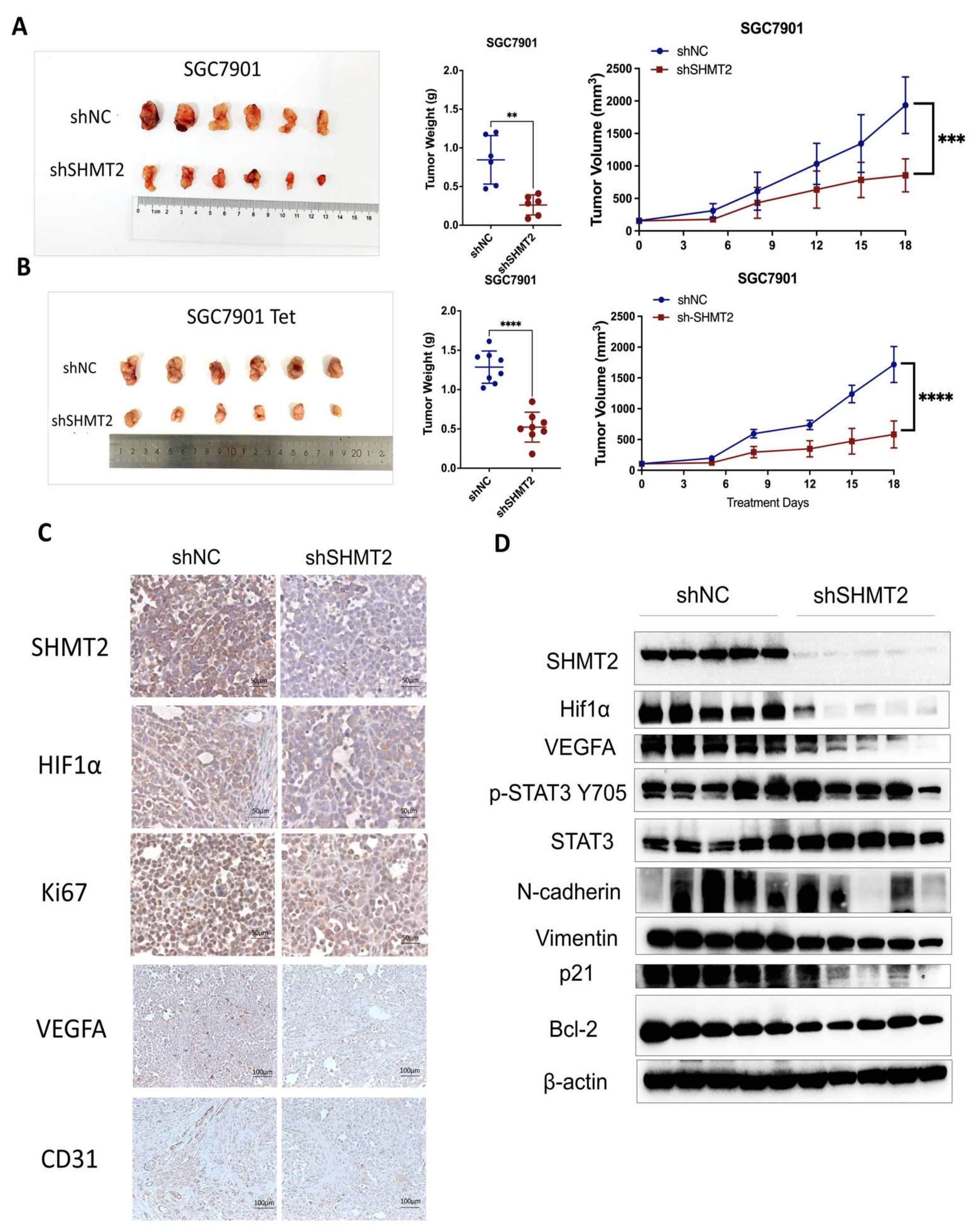
2.7. SHMT2 Knockdown Suppressed the Tumorigenesis of GC Cells In Vivo
3. Discussion
4. Materials and Methods
4.1. Analyses Based on Public Databases
4.2. Cell Culture and Reagents
4.3. Lentiviral Packaging and Virus Infection
4.4. Western Blot Analysis
4.5. Real-Time Quantitative PCR
4.6. CCK-8 Cell Viability Assay
4.7. Cell Cycle and Cell Apoptosis
4.8. EdU Assay
4.9. Wound-Healing Assay and Transwell Assay
4.10. Clonogenic Assay and Soft Agar Colony Formation Assay
4.11. Seahorse XF Glycolysis Assay
4.12. ROS Assay and GSH Assay
4.13. Animal Experiments
4.14. Immunohistochemical Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhao, Y.; Zhou, Y.; Tian, Y.; He, Q.; Lin, J.; Hao, H.; Zou, B.; Jiang, L.; Zhao, G.; et al. Comparison of Survival and Patterns of Recurrence in Gastric Neuroendocrine Carcinoma, Mixed Adenoneuroendocrine Carcinoma, and Adenocarcinoma. JAMA Netw. Open 2021, 4, e2114180. [Google Scholar] [CrossRef]
- Li, K.; Zhang, A.; Li, X.; Zhang, H.; Zhao, L. Advances in clinical immunotherapy for gastric cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188615. [Google Scholar] [CrossRef]
- Digklia, A.; Wagner, A.D. Advanced gastric cancer: Current treatment landscape and future perspectives. World J. Gastroenterol. 2016, 22, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.; Ravelli, A.; Polom, K.; Petrioli, R.; Marano, L.; Marrelli, D.; Roviello, F.; Generali, D. Apatinib: A novel receptor tyrosine kinase inhibitor for the treatment of gastric cancer. Cancer Lett. 2016, 372, 187–191. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, S.Y.; Hwang, J.A.; Hong, S.H.; Shin, A.; Choi, I.J.; Lee, Y.S. Association Study between Folate Pathway Gene Single Nucleotide Polymorphisms and Gastric Cancer in Koreans. Genom. Inform. 2012, 10, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Woo, H.D.; Lee, J.; Choi, I.J.; Kim, Y.W.; Sung, J.; Kim, J. Dietary folate, one-carbon metabolism-related genes, and gastric cancer risk in Korea. Mol. Nutr. Food Res. 2016, 60, 337–345. [Google Scholar] [CrossRef]
- Kweon, S.S.; Shu, X.O.; Xiang, Y.; Yang, G.; Ji, B.T.; Li, H.; Gao, Y.T.; Zheng, W.; Shrubsole, M.J. One-carbon metabolism dietary factors and distal gastric cancer risk in chinese women. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1374–1382. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Wang, X.; Jian, H.; Xiao, H.; Wen, T. SHMT2 promotes cell viability and inhibits ROS-dependent, mitochondrial-mediated apoptosis via the intrinsic signaling pathway in bladder cancer cells. Cancer Gene Ther. 2022, 29, 1514–1527. [Google Scholar] [CrossRef]
- Wei, Z.; Song, J.; Wang, G.; Cui, X.; Zheng, J.; Tang, Y.; Chen, X.; Li, J.; Cui, L.; Liu, C.Y.; et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 2018, 9, 4468. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef]
- Wang, H.; Chong, T.; Li, B.Y.; Chen, X.S.; Zhen, W.B. Evaluating the clinical significance of SHMT2 and its co-expressed gene in human kidney cancer. Biol. Res. 2020, 53, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, Q. Overexpression of SHMT2 Predicts a Poor Prognosis and Promotes Tumor Cell Growth in Bladder Cancer. Front. Genet. 2021, 12, 682856. [Google Scholar] [CrossRef]
- Jin, Y.; Jung, S.-N.; Lim, M.A.; Oh, C.; Piao, Y.; Kim, H.J.; Nguyena, Q.; Kang, Y.E.; Chang, J.W.; Won, H.-R.; et al. SHMT2 Induces Stemness and Progression of Head and Neck Cancer. Int. J. Mol. Sci. 2022, 23, 9714. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Fang, X.; Li, Y.; Zhang, Y. High Expression of Serine Hydroxymethyltransferase 2 Indicates Poor Prognosis of Gastric Cancer Patients. Med. Sci. Monit. 2019, 25, 7430–7438. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, T. Knockdown of SHMT2 enhances the sensitivity of gastric cancer cells to radiotherapy through the Wnt/β-catenin pathway. Open Life Sci. 2022, 17, 1249–1255. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef]
- Wang, J.Z.; Zhu, W.; Han, J.; Yang, X.; Zhou, R.; Lu, H.C.; Yu, H.; Yuan, W.B.; Li, P.C.; Tao, J.; et al. The role of the HIF-1α/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun. 2021, 41, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Khromova, N.V.; Kopnin, P.B.; Stepanova, E.V.; Agapova, L.S.; Kopnin, B.P. p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K.; et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621.e615. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Hanson, R.W. Resurgence of serine: An often neglected but indispensable amino Acid. J. Biol. Chem. 2012, 287, 19786–19791. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Wu, K.J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef] [PubMed]
- Donato, C.; Kunz, L.; Castro-Giner, F.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O.; et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Rubini, E.; Paglia, G.; Chichiarelli, S.; Giamogante, F.; Macone, A.; Perugia, G.; Magliocca, F.M.; Gurtner, A.; et al. Shmt2: A Stat3 Signaling New Player in Prostate Cancer Energy Metabolism. Cells 2019, 8, 1048. [Google Scholar] [CrossRef]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef]
- Xia, J.; Zhang, J.; Wu, X.; Du, W.; Zhu, Y.; Liu, X.; Liu, Z.; Meng, B.; Guo, J.; Yang, Q.; et al. Blocking glycine utilization inhibits multiple myeloma progression by disrupting glutathione balance. Nat. Commun. 2022, 13, 4007. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef]
- Tiwari, V.; Daoud, E.V.; Hatanpaa, K.J.; Gao, A.; Zhang, S.; An, Z.; Ganji, S.K.; Raisanen, J.M.; Lewis, C.M.; Askari, P.; et al. Glycine by MR spectroscopy is an imaging biomarker of glioma aggressiveness. Neuro Oncol. 2020, 22, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, L.; Liu, X.; Tan, Y.; Tao, L.; Xiao, Y.; Deng, P.; Wang, H.; Deng, Q.; Lin, Y.; et al. Cytoplasmic SHMT2 drives the progression and metastasis of colorectal cancer by inhibiting β-catenin degradation. Theranostics 2021, 11, 2966–2986. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, M.; Du, T.; Hou, Z.; You, S.; Zhang, S.; Ji, M.; Xue, N.; Chen, X. SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling. Int. J. Mol. Sci. 2023, 24, 7150. https://doi.org/10.3390/ijms24087150
Wang W, Wang M, Du T, Hou Z, You S, Zhang S, Ji M, Xue N, Chen X. SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling. International Journal of Molecular Sciences. 2023; 24(8):7150. https://doi.org/10.3390/ijms24087150
Chicago/Turabian StyleWang, Weida, Mingjin Wang, Tingting Du, Zhenyan Hou, Shen You, Sen Zhang, Ming Ji, Nina Xue, and Xiaoguang Chen. 2023. "SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling" International Journal of Molecular Sciences 24, no. 8: 7150. https://doi.org/10.3390/ijms24087150
APA StyleWang, W., Wang, M., Du, T., Hou, Z., You, S., Zhang, S., Ji, M., Xue, N., & Chen, X. (2023). SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling. International Journal of Molecular Sciences, 24(8), 7150. https://doi.org/10.3390/ijms24087150