

Genome-Wide Association Study of Age at First Calving in U.S. Holstein Cows


Abstract
1. Introduction
2. Results and Discussion
2.1. Additive Effects
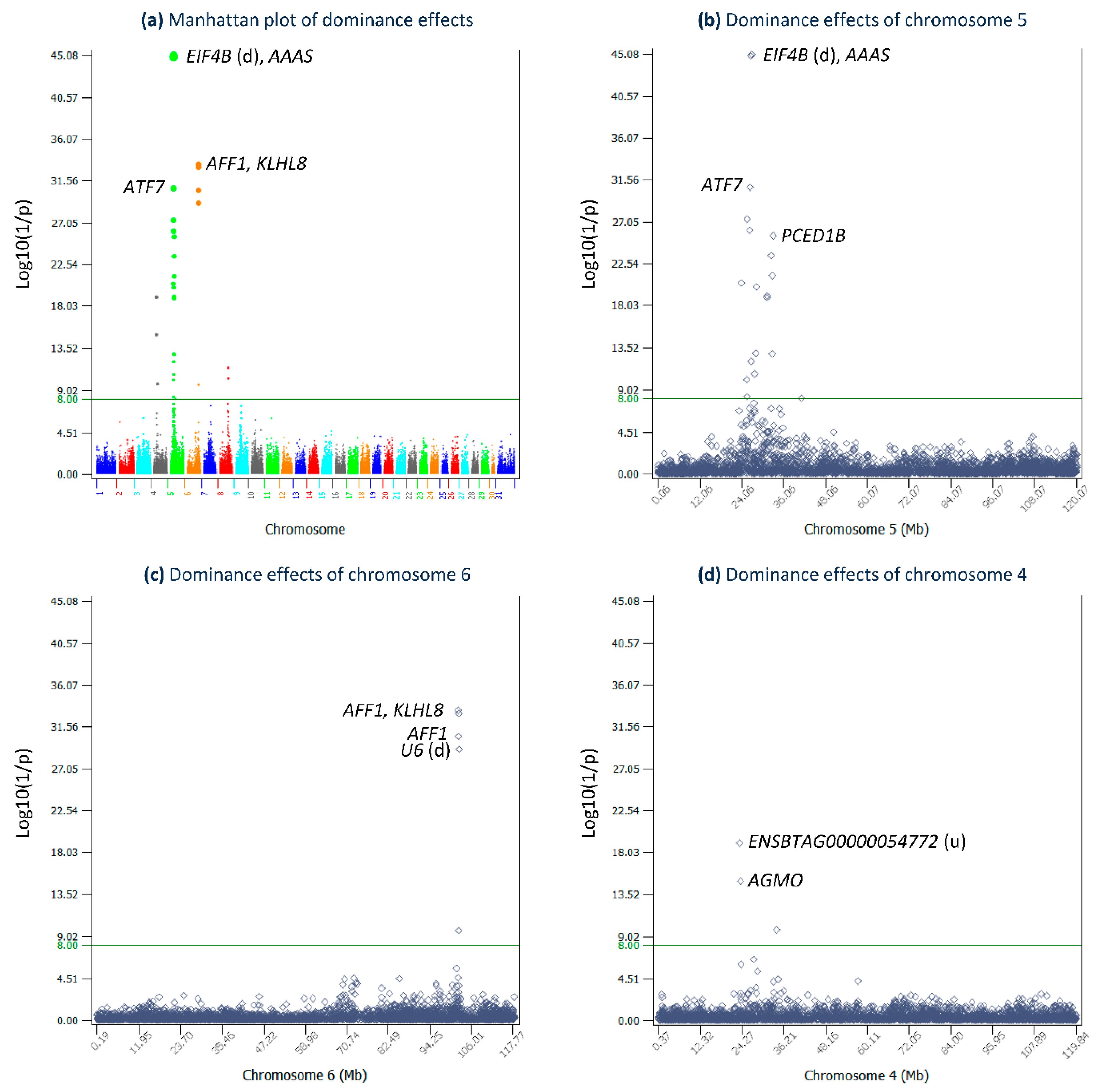
2.2. Dominance Effects
2.3. Elimination of Rare Negative Recessive Genotypes for Heifer Culling
2.4. Comparison with Previous Studies
2.5. Gene Ontology of Candidate Genes
3. Materials and Methods
3.1. Holstein Population and SNP Data
3.2. GWAS Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchison, J.; VanRaden, P.; Null, D.; Cole, J.; Bickhart, D. Genomic evaluation of age at first calving. J. Dairy Sci. 2017, 100, 6853–6861. [Google Scholar] [CrossRef] [PubMed]
- Norman, D.; Hutchison, J. New Trait: Early First Calving. 2019. Available online: https://queries.uscdcb.com/News/CDCB%20Connection%20Early%20First%20Calving%2003_2019.pdf (accessed on 10 April 2023).
- Ma, L.; Cole, J.; Da, Y.; VanRaden, P. Symposium review: Genetics, genome-wide association study, and genetic improvement of dairy fertility traits. J. Dairy Sci. 2018, 102, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, L.; Prakapenka, D.; VanRaden, P.M.; Cole, J.B.; Da, Y. A large-scale genome-wide association study in US Holstein cattle. Front. Genet. 2019, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- The National Center for Biotechnology Information. PGR Progesterone Receptor. Available online: https://www.ncbi.nlm.nih.gov/gene/5241 (accessed on 10 April 2023).
- The National Center for Biotechnology Information. SHBG Sex Hormone Binding Globulin. Available online: https://www.ncbi.nlm.nih.gov/gene/6462 (accessed on 10 April 2023).
- Aydın, B.; Winters, S.J. Sex hormone-binding globulin in children and adolescents. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Violetis, O.; Chatzakis, C.; Triantafyllidou, O.; Eleftheriades, M.; Lambrinoudaki, I.; Mastorakos, G.; Vlahos, N.F. Daughters of polycystic ovary syndrome pregnancies and androgen levels in puberty: A Meta-analysis. Gynecol. Endocrinol. 2022, 38, 822–830. [Google Scholar] [CrossRef] [PubMed]
- The National Center for Biotechnology Information. HS3ST3A1 Heparan Sulfate-Glucosamine 3-Sulfotransferase 3A1. Available online: https://www.ncbi.nlm.nih.gov/gene/9955 (accessed on 10 April 2023).
- Ramirez-Diaz, J.; Cenadelli, S.; Bornaghi, V.; Bongioni, G.; Montedoro, S.; Achilli, A.; Capelli, C.; Rincon, J.; Milanesi, M.; Passamonti, M.M. Identification of genomic regions associated with total and progressive sperm motility in Italian Holstein bulls. J. Dairy Sci. 2023, 106, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Davies, C. Why is the fetal allograft not rejected? J. Anim. Sci. 2007, 85, E32–E35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Schenkel, F.S.; Melo, A.L.; Oliveira, H.R.; Pedrosa, V.B.; Araujo, A.C.; Melka, M.G.; Brito, L.F. Identifying pleiotropic variants and candidate genes for fertility and reproduction traits in Holstein cattle via association studies based on imputed whole-genome sequence genotypes. BMC Genom. 2022, 23, 331. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Júnior, G.A.; de Oliveira, H.N.; Carvalheiro, R.; Cardoso, D.F.; Fonseca, L.F.S.; Ventura, R.V.; de Albuquerque, L.G. Whole-genome sequencing provides new insights into genetic mechanisms of tropical adaptation in Nellore (Bos primigenius indicus). Sci. Rep. 2020, 10, 9412. [Google Scholar] [CrossRef] [PubMed]
- Strucken, E.M.; Bortfeldt, R.H.; Tetens, J.; Thaller, G.; Brockmann, G.A. Genetic effects and correlations between production and fertility traits and their dependency on the lactation-stage in Holstein Friesians. BMC Genet. 2012, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Mota, L.F.; Lopes, F.B.; Fernandes Júnior, G.A.; Rosa, G.J.; Magalhães, A.F.; Carvalheiro, R.; Albuquerque, L.G. Genome-wide scan highlights the role of candidate genes on phenotypic plasticity for age at first calving in Nellore heifers. Sci. Rep. 2020, 10, 6481. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Guo, G.; Wang, Y.; Guo, X.; Zhang, X.; Liu, L.; Shi, W.; Li, X.; Su, G.; Zhang, Q. Genetic analysis and genome wide association studies for age at first calving in Chinese Holsteins. Acta Vet. Zootech. Sin. 2015, 46, 373–381. [Google Scholar]
- The Gene Ontology Resources. Available online: http://geneontology.org/ (accessed on 10 April 2023).
- KEGG Pathway Database. Available online: https://www.genome.jp/kegg/pathway.html (accessed on 10 April 2023).
- DAVID Bioinformatics Resources. Available online: https://david.ncifcrf.gov/ (accessed on 10 April 2023).
- Ma, L.; Runesha, H.B.; Dvorkin, D.; Garbe, J.; Da, Y. Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies. BMC Bioinform. 2008, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Weeks, N.T.; Luecke, G.R.; Groth, B.M.; Kraeva, M.; Ma, L.; Kramer, L.M.; Koltes, J.E.; Reecy, J.M. High-performance epistasis detection in quantitative trait GWAS. Int. J. High Perform. Comput. Appl. 2016, 32, 321–336. [Google Scholar] [CrossRef]
- Henderson, C. Applications of Linear Models in Animal Breeding; University of Guelph: Guelph, ON, Canada, 1984. [Google Scholar]
- Mao, Y.; London, N.R.; Ma, L.; Dvorkin, D.; Da, Y. Detection of SNP epistasis effects of quantitative traits using an extended Kempthorne model. Physiol. Genom. 2006, 28, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics, 4th ed.; Longmans Green: Harlow, UK, 1996. [Google Scholar]
- Wang, S.; Dvorkin, D.; Da, Y. SNPEVG: A graphical tool for GWAS graphing with mouse clicks. BMC Bioinform. 2012, 13, 319. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| SNP | Chr | Position (bp) | Candidate Gene | Effect (α, −Days) | al+ | ae+ (−Days) | f_al+ | al− | ae− (−Days) | f_al− | log10(1/p) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs110401500 | 19 | 31,252,963 | ARHGAP44 | −1.02 | 2 | 0.730 | 0.287 | 1 | −0.294 | 0.713 | 37.30 |
| rs41257332 | 19 | 33,443,229 | TTC19 | −0.97 | 2 | 0.616 | 0.366 | 1 | −0.355 | 0.634 | 36.45 |
| rs111004845 | 19 | 27,355,811 | SHBG (9664 bp u) a | −0.97 | 2 | 0.648 | 0.332 | 1 | −0.323 | 0.668 | 36.06 |
| rs135712994 | 19 | 33,421,057 | NCOR1 | −0.89 | 2 | 0.359 | 0.597 | 1 | −0.531 | 0.403 | 33.14 |
| rs110761858 | 19 | 33,358,794 | NCOR1 | −0.87 | 2 | 0.354 | 0.592 | 1 | −0.514 | 0.408 | 31.87 |
| rs41621822 | 19 | 31,902,307 | LOC112442639 | −0.90 | 2 | 0.594 | 0.339 | 1 | −0.304 | 0.661 | 31.39 |
| rs133729181 | 19 | 32,106,657 | COX10 | 0.86 | 1 | 0.519 | 0.394 | 2 | −0.337 | 0.606 | 30.40 |
| rs134054295 | 23 | 32,599,962 | LOC537017 | −1.02 | 2 | 0.808 | 0.211 | 1 | −0.217 | 0.789 | 30.30 |
| rs136368496 | 23 | 28,526,405 | LOC101905956 | −0.87 | 2 | 0.555 | 0.364 | 1 | −0.318 | 0.636 | 30.04 |
| rs110845473 | 19 | 27,484,633 | DNAH2 | −0.98 | 2 | 0.753 | 0.234 | 1 | −0.230 | 0.766 | 29.89 |
| rs41904669 | 19 | 27,073,319 | TNK1 | 0.97 | 1 | 0.738 | 0.239 | 2 | −0.232 | 0.761 | 29.43 |
| rs109836072 | 15 | 7,475,196 | TRPC6 (3151 bp d) a | −1.04 | 2 | 0.818 | 0.212 | 1 | −0.220 | 0.788 | 29.38 |
| rs137457305 | 23 | 26,926,436 | C23H6orf10 | 0.98 | 1 | 0.746 | 0.236 | 2 | −0.231 | 0.764 | 29.28 |
| rs41904556 | 19 | 27,316,118 | MPDU1 | 0.96 | 1 | 0.732 | 0.240 | 2 | −0.231 | 0.760 | 29.03 |
| rs42688274 | 19 | 29,273,714 | GAST (22,315 bp d) | −0.97 | 2 | 0.740 | 0.235 | 1 | −0.227 | 0.765 | 29.03 |
| rs29010491 | 23 | 30,176,828 | ENSBTAG00000051232 (21,566 bp u) a | 0.81 | 1 | 0.442 | 0.453 | 2 | −0.366 | 0.547 | 28.03 |
| rs137317833 | 23 | 29,958,908 | blank | −0.81 | 2 | 0.455 | 0.441 | 1 | −0.358 | 0.559 | 27.93 |
| rs136764006 | 15 | 7,861,416 | PGR | 0.89 | 1 | 0.620 | 0.303 | 2 | −0.270 | 0.697 | 27.63 |
| rs110654893 | 23 | 30,013,004 | ZNF311 (5017 bp u) a | −0.80 | 2 | 0.438 | 0.453 | 1 | −0.363 | 0.547 | 27.53 |
| rs109681200 | 23 | 30,377,501 | ZSCAN31 | −0.83 | 2 | 0.538 | 0.354 | 1 | −0.294 | 0.646 | 27.39 |
| SNP | Chr | Position | Candidate Gene | Effect (δ, −Days) | DR | d_DR (−Days) | f_DR | DD | d_DD (−Days) | f_DD | RR | d_RR (−Days) | f_RR | f_R | log10(1/p) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs109438971 | 5 | 26,964,045 | EIF4B (14,636 bp d) | 5.53 | 12 | 0.62 | 0.152 | 22 | −0.06 | 0.843 | 11 | −9.76 | 0.005 | 0.081 | 45.08 |
| rs110558219 | 5 | 26,715,326 | AAAS | 5.51 | 12 | 0.62 | 0.152 | 11 | −0.06 | 0.843 | 22 | −9.72 | 0.005 | 0.081 | 44.89 |
| rs43768813 | 6 | 101,887,271 | AFF1 | 4.87 | 12 | 0.61 | 0.131 | 22 | −0.05 | 0.864 | 11 | −8.48 | 0.005 | 0.07 | 33.36 |
| rs42739334 | 6 | 102,065,812 | KLHL8 | 4.68 | 12 | 0.60 | 0.136 | 11 | −0.05 | 0.859 | 22 | −8.11 | 0.005 | 0.073 | 33.04 |
| rs109675908 | 5 | 26,499,453 | ATF7 | 3.88 | 12 | 0.54 | 0.170 | 22 | −0.05 | 0.823 | 11 | −6.63 | 0.007 | 0.092 | 30.77 |
| rs43480825 | 6 | 101,994,654 | AFF1 | 4.57 | 12 | 0.57 | 0.135 | 11 | −0.04 | 0.860 | 22 | −7.95 | 0.005 | 0.072 | 30.56 |
| rs109933750 | 6 | 102,164,971 | U6 (16,972 bp d) | 4.50 | 12 | 0.56 | 0.134 | 22 | −0.04 | 0.861 | 11 | −7.83 | 0.005 | 0.072 | 29.18 |
| rs135494774 | 5 | 25,556,149 | NCKAP1L | 3.46 | 12 | 0.53 | 0.180 | 11 | −0.06 | 0.811 | 22 | −5.80 | 0.008 | 0.099 | 27.34 |
| rs134764130 | 5 | 26,385,947 | ATP5MC2 (7720 bp d) | 3.14 | 12 | 0.51 | 0.192 | 22 | −0.06 | 0.798 | 11 | −5.18 | 0.010 | 0.106 | 26.13 |
| rs41603412 | 5 | 33,076,713 | PCED1B | 3.17 | 12 | 0.50 | 0.185 | 22 | −0.06 | 0.806 | 11 | −5.28 | 0.009 | 0.101 | 25.56 |
| SNP | Formula of Negative Impact a | AFC (Days) | Milk Yield (kg) | Fat Yield (kg) | Protein Yield (kg) |
|---|---|---|---|---|---|
| rs109675908 | b | 7.69 | −470.06 | −20.14 | −14.22 |
| rs110558219 | 10.75 | −646.33 | −26.03 | −19.27 | |
| rs109438971 | 10.88 | −640.94 | −26.03 | −19.11 | |
| rs43768813 | 12.50 | −169.35 | −9.05 | −5.73 | |
| rs43480825 | 12.00 | −189.78 | −9.88 | −6.40 | |
| rs42739334 | 12.83 | −240.84 | −10.50 | −7.41 | |
| rs109933750 | 11.76 | −201.23 | −10.22 | −6.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prakapenka, D.; Liang, Z.; Da, Y. Genome-Wide Association Study of Age at First Calving in U.S. Holstein Cows. Int. J. Mol. Sci. 2023, 24, 7109. https://doi.org/10.3390/ijms24087109
Prakapenka D, Liang Z, Da Y. Genome-Wide Association Study of Age at First Calving in U.S. Holstein Cows. International Journal of Molecular Sciences. 2023; 24(8):7109. https://doi.org/10.3390/ijms24087109
Chicago/Turabian StylePrakapenka, Dzianis, Zuoxiang Liang, and Yang Da. 2023. "Genome-Wide Association Study of Age at First Calving in U.S. Holstein Cows" International Journal of Molecular Sciences 24, no. 8: 7109. https://doi.org/10.3390/ijms24087109
APA StylePrakapenka, D., Liang, Z., & Da, Y. (2023). Genome-Wide Association Study of Age at First Calving in U.S. Holstein Cows. International Journal of Molecular Sciences, 24(8), 7109. https://doi.org/10.3390/ijms24087109