
Dynamics of the Equine Placental DNA Methylome and Transcriptome from Mid- to Late Gestation

 ,
,  ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
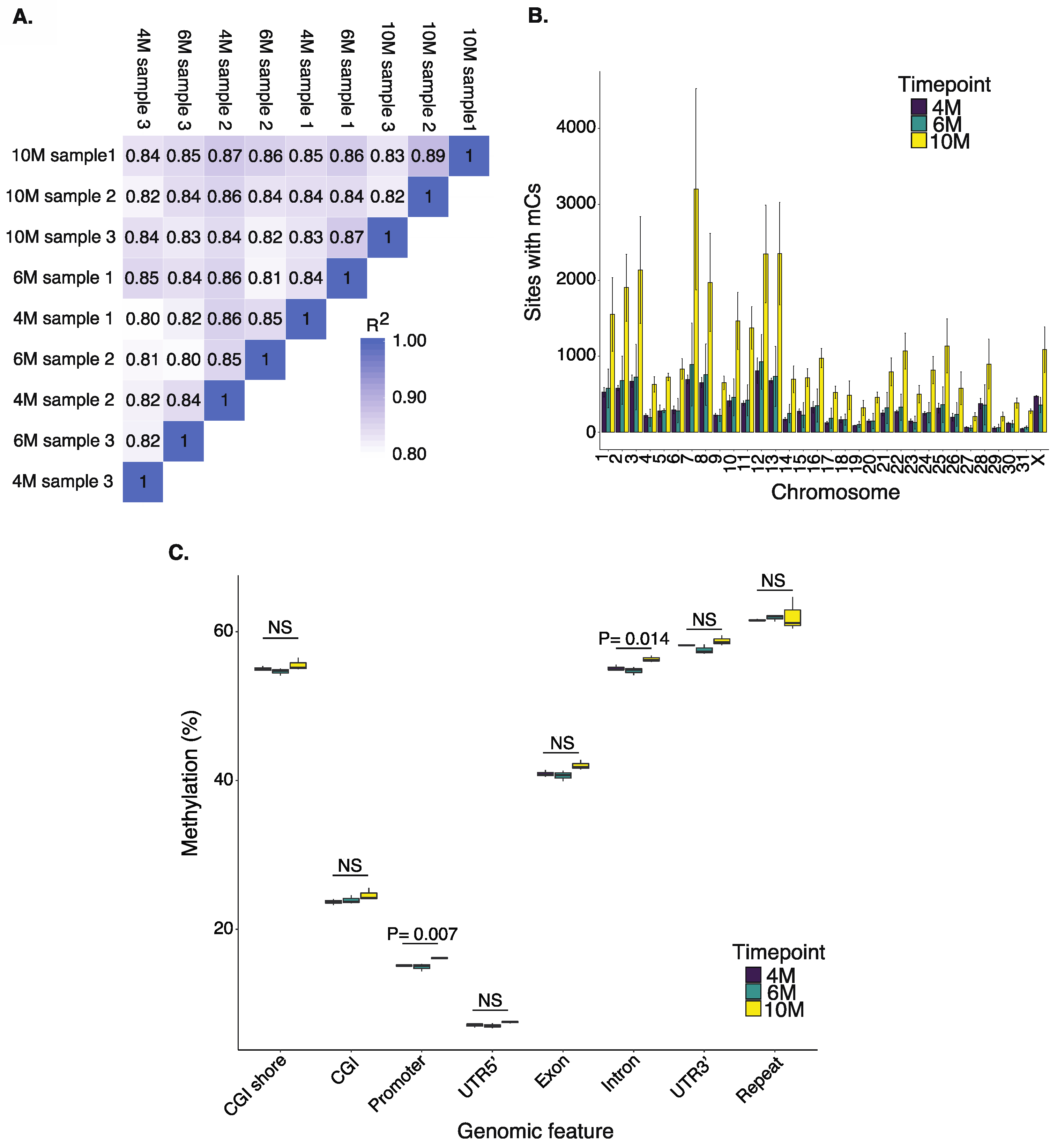
2.1. Methylation Patterns in the Equine Placenta
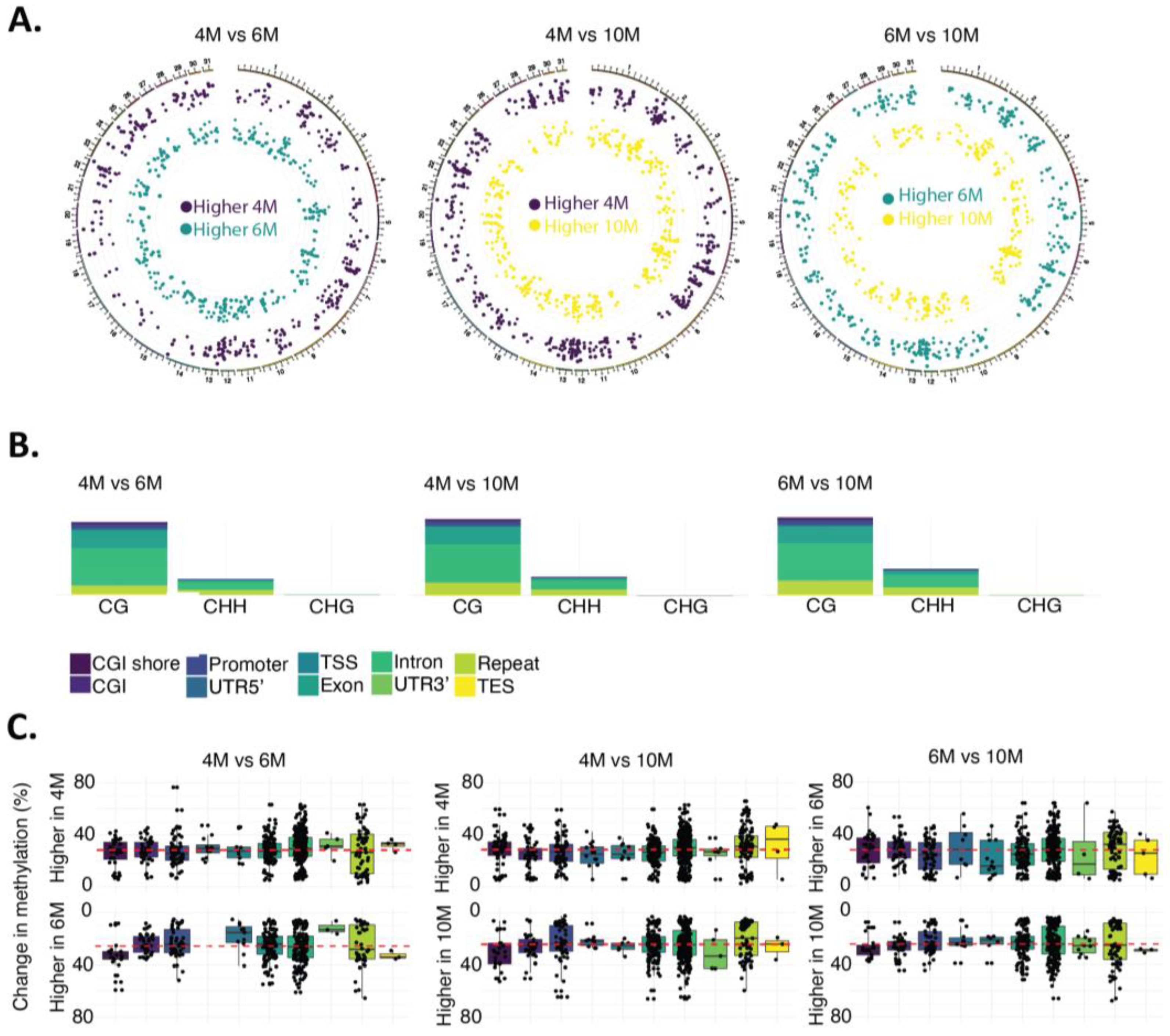
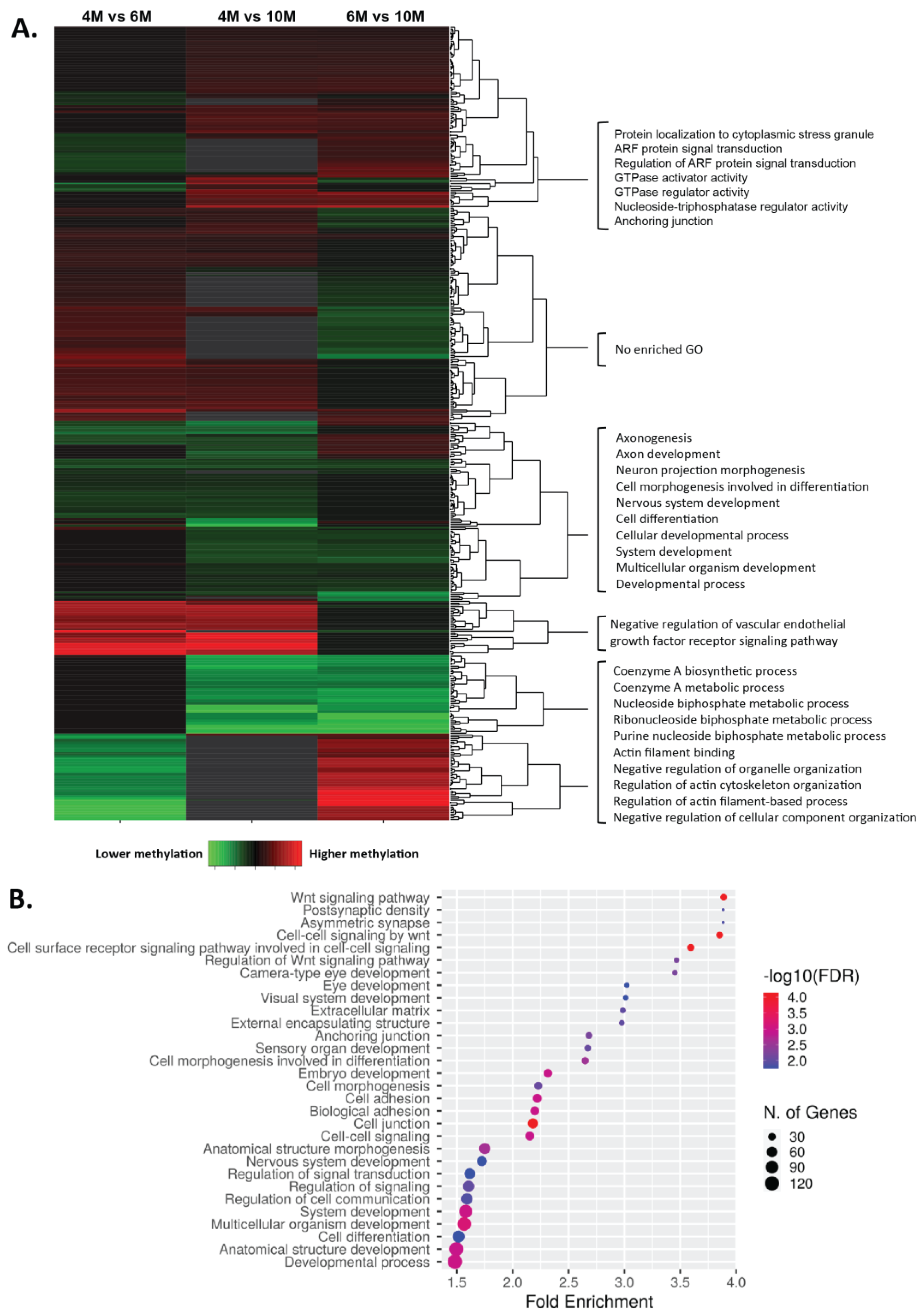
2.2. Dynamics of DNA Methylation during Gestation
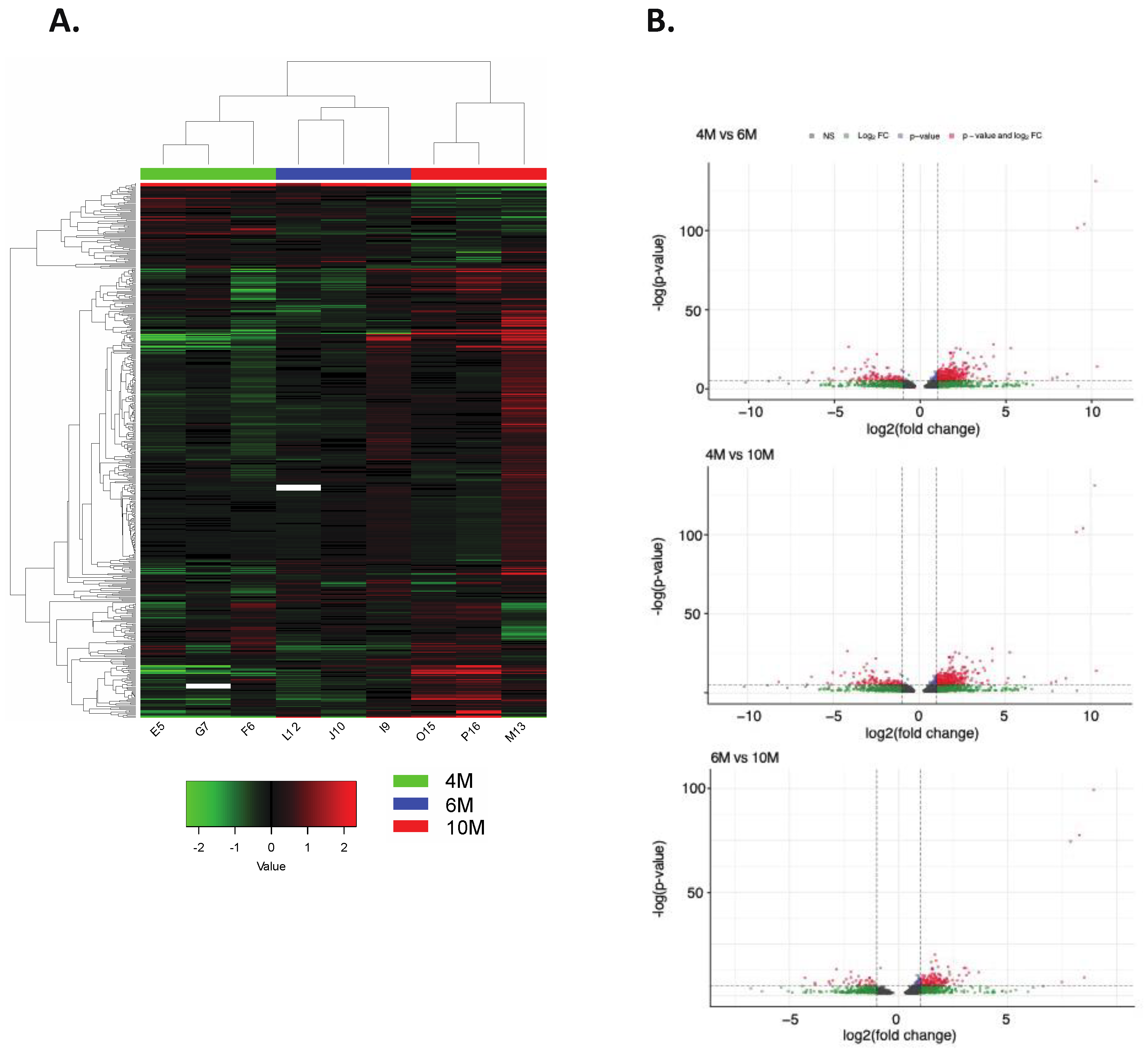
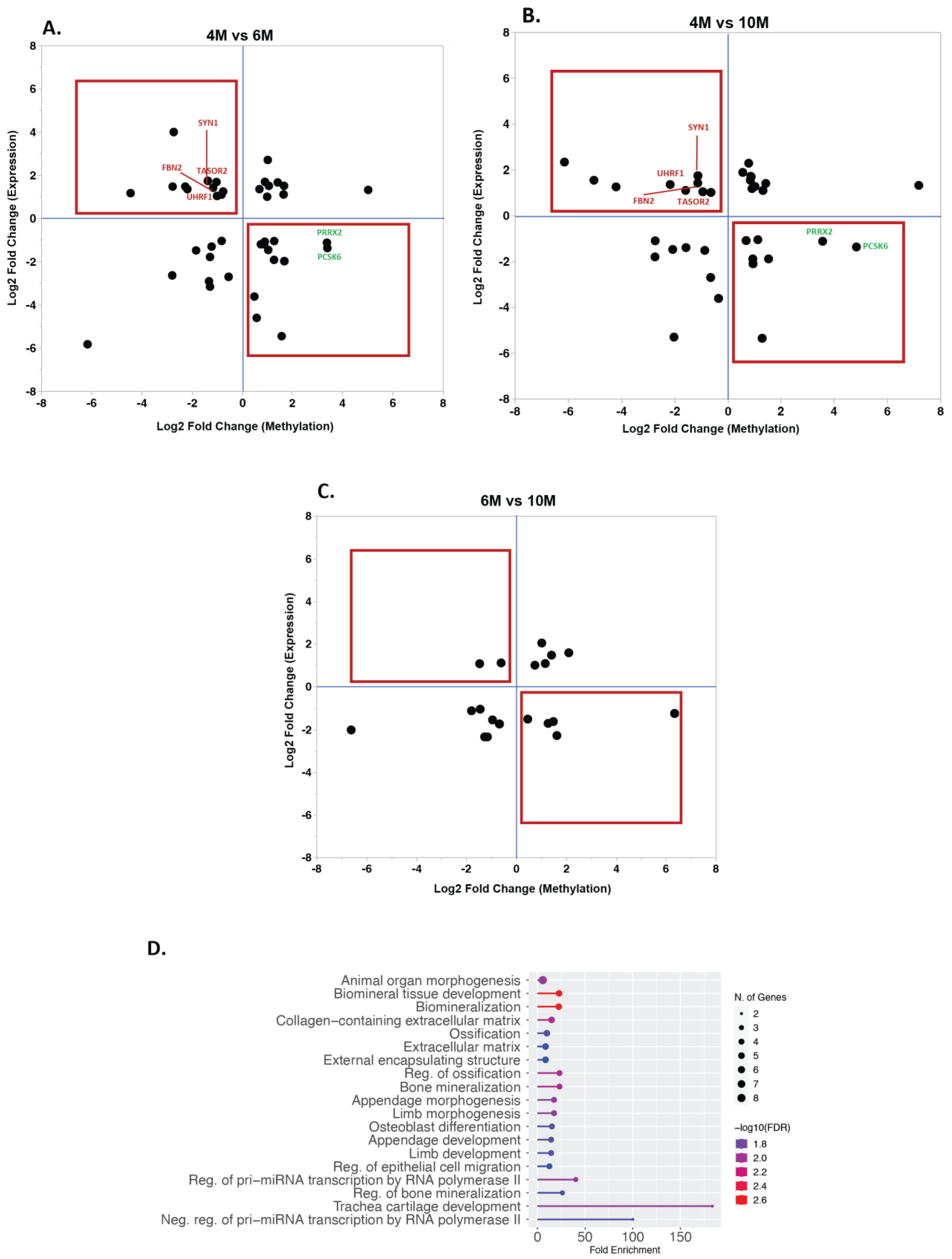
2.3. Impact of Placental DNA Methylation on Gene Expression
3. Discussion
4. Materials and Methods
4.1. Animal Use and Sample Collection
4.2. DNA Methylome Analysis
4.2.1. DNA Extraction, Library Preparation, and Sequencing
4.2.2. Data Processing and Bioinformatic Analysis
4.3. Transcriptome Analysis
4.3.1. RNA Extraction, Library Preparation, and Sequencing
4.3.2. Data Processing and Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef]
- Adamson, S.L.; Lu, Y.; Whiteley, K.J.; Holmyard, D.; Hemberger, M.; Pfarrer, C.; Cross, J.C. Interactions between Trophoblast Cells and the Maternal and Fetal Circulation in the Mouse Placenta. Dev. Biol. 2002, 250, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Koukoura, O.; Sifakis, S.; Spandidos, D.A. DNA methylation in the human placenta and fetal growth. Mol. Med. Rep. 2012, 5, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Yuen, R.K.; Gordon, L.; Penaherrera, M.S.; Sharkey, A.; Moffett, A.; Craig, J.M.; Robinson, W.P.; Saffery, R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genom. 2011, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, C.P.; Goyal, R.; Mittal, A.; Longo, L.D. Gene Expression in the Placenta. Int. J. Dev. Biol. 2010, 54, 507–523. [Google Scholar] [CrossRef]
- Ng, H.K.; Novakovic, B.; Hiendleder, S.; Craig, J.M.; Roberts, C.T.; Saffery, R. Distinct Patterns of Gene-Specific Methylation in Mammalian Placentas: Implications for Placental Evolution and Function. Placenta 2010, 31, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Turner, W. Some General Observations on the Placenta, with especial reference to the Theory of Evolution. J. Anat. Physiol. 1876, 11, 33–53. [Google Scholar] [PubMed]
- Dini, P.; Kalbfleisch, T.; Uribe-Salazar, J.M.; Carossino, M.; Ali, H.E.-S.; Loux, S.C.; Esteller-Vico, A.; Norris, J.K.; Anand, L.; Scoggin, K.E.; et al. Parental bias in expression and interaction of genes in the equine placenta. Proc. Natl. Acad. Sci. USA 2021, 118, e2006474118. [Google Scholar] [CrossRef]
- Carter, A.M.; Enders, A.C. The evolution of epitheliochorial placentation. Annu. Rev. Anim. Biosci. 2013, 1, 443–467. [Google Scholar] [CrossRef]
- Dini, P.; Norris, J.; Ali, H.E.S.; Loux, S.C.; Carossino, M.; Esteller-Vico, A.; Bailey, E.; Kalbfleisch, T.; Daels, P.; Ball, B.A. Landscape of overlapping gene expression in the equine placenta. Genes 2019, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Loux, S.C.; Dini, P.; Ali, H.E.S.; Kalbfleisch, T.; Ball, B.A. Characterization of the placental transcriptome through mid to late gestation in the mare. PLoS ONE 2019, 14, e0224497. [Google Scholar] [CrossRef]
- Carter, A.M.; Enders, A.C. Comparative aspects of trophoblast development and placentation. Reprod. Biol. Endocrinol. 2004, 2, 46. [Google Scholar] [CrossRef]
- Proudhon, C.; Bourc’his, D. Identification and resolution of artifacts in the interpretation of imprinted gene expression. Brief. Funct. Genom. 2010, 9, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Ginther, O. Reproductive Biology of the Mare; Equiservices: Cross Plains, WI, USA, 1992. [Google Scholar]
- Allen, W.; Stewart, F. Equine Chorionic Gonadotropin. In Equine Reproduction; Wiley-Blackwell: Hoboken, NJ, USA, 1993. [Google Scholar]
- Conley, A.J. Review of the reproductive endocrinology of the pregnant and parturient mare. Theriogenology 2016, 86, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Holtan, D.; Squires, E.; Lapin, D.; Ginther, O. Effect of ovariectomy on pregnancy in mares. J. Reprod. Fertil. Suppl. 1979, 27, 457–463. [Google Scholar]
- Loux, S.C.; Conley, A.J.; Scoggin, K.E.; El-Sheikh Ali, H.; Dini, P.; Ball, B.A. New insights in equine steroidogenesis: An in-depth look at steroid signaling in the placenta. Reproduction 2020, 160, 65–82. [Google Scholar] [CrossRef]
- Dini, P.; Daels, P.; Loux, S.C.; Esteller-Vico, A.; Carossino, M.; Scoggin, K.E.; Ball, B.A. Kinetics of the chromosome 14 microRNA cluster ortholog and its potential role during placental development in the pregnant mare. BMC Genom. 2018, 19, 954. [Google Scholar] [CrossRef]
- El-Sheikh Ali, H.; Legacki, E.L.; Scoggin, K.E.; Loux, S.C.; Dini, P.; Esteller-Vico, A.; Conley, A.J.; Stanley, S.D.; Ball, B.A. Steroid synthesis and metabolism in the equine placenta during placentitis. Reproduction 2020, 159, 289–302. [Google Scholar] [CrossRef]
- Dini, P.; Ali, H.E.S.; Carossino, M.; Loux, S.C.; Esteller-Vico, A.; Scoggin, K.E.; Daels, P.; Ball, B.A. Expression profile of the chromosome 14 microRNA cluster (C14MC) ortholog in equine maternal circulation throughout pregnancy and its potential implications. Int. J. Mol. Sci. 2019, 20, 6285. [Google Scholar] [CrossRef]
- Siegfried, Z.; Simon, I. DNA methylation and gene expression. Wiley Interdiscip. Rev. Syst Biol. Med. 2010, 2, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Grigoriu, A.; Ferreira, J.C.; Choufani, S.; Baczyk, D.; Kingdom, J.; Weksberg, R. Cell specific patterns of methylation in the human placenta. Epigenetics 2011, 6, 368–379. [Google Scholar] [CrossRef]
- Lee, J.R.; Hong, C.P.; Moon, J.W.; Jung, Y.D.; Kim, D.S.; Kim, T.H.; Gim, J.A.; Bae, J.H.; Choi, Y.; Eo, J.; et al. Genome-wide analysis of DNA methylation patterns in horse. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Robinson, W.P.; Price, E.M. The human placental methylome. Cold Spring Harb. Perspect. Med. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Mayne, B.T.; Buckberry, S.; Breen, J.; Rodriguez Lopez, C.M.; Roberts, C.T. Recent progress towards understanding the role of DNA methylation in human placental development. Reproduction 2016, 152, R23–R30. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.K.; Hong, D.; Crary, F.; Ashwood, P.; Walker, C.; Korf, I.; Robinson, W.P.; et al. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Novakovic, B.; Saffery, R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics 2010, 2, 627–638. [Google Scholar] [CrossRef]
- Vlahos, A.; Mansell, T.; Saffery, R.; Novakovic, B. Human placental methylome in the interplay of adverse placental health, environmental exposure, and pregnancy outcome. PLoS Genet. 2019, 15, e1008236. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, M. DNA Methylation and Complex Human Disease; Academic Press: Cambridge, MA, USA, 2015; ISBN 0127999205. [Google Scholar]
- Gamage, T.K.J.B.; Schierding, W.; Hurley, D.; Tsai, P.; Ludgate, J.L.; Bhoothpur, C.; Chamley, L.W.; Weeks, R.J.; Macaulay, E.C.; James, J.L. The role of DNA methylation in human trophoblast differentiation. Epigenetics 2018, 13, 1154–1173. [Google Scholar] [CrossRef] [PubMed]
- Falick Michaeli, T.; Spiro, A.; Sabag, O.; Karavani, G.; Yagel, S.; Eventov-Friedman, S.; Cedar, H.; Bergman, Y.; Gielchinsky, Y. Determining gestational age using genome methylation profile: A novel approach for fetal medicine. Prenat. Diagn. 2019, 39, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Pelizzola, M.; Ecker, J.R. The DNA Methylome. FEBS Lett. 2011, 585, 1994–2000. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Fisher, S.J. Placenta: The Forgotten Organ. Annu. Rev. Cell Dev. Biol. 2015, 31, 523–552. [Google Scholar] [CrossRef]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef]
- Winn, V.D.; Haimov-Kochman, R.; Paquet, A.C.; Yang, Y.J.; Madhusudhan, M.S.; Gormley, M.; Feng, K.T.V.; Bernlohr, D.A.; McDonagh, S.; Pereira, L.; et al. Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 2007, 148, 1059–1079. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Nabekura, T.; Kaddoumi, A.; Bammler, T.K.; Govindarajan, R.; Hebert, M.F.; Unadkat, J.D. Profiling gene expression in human placentae of different gestational ages: An OPRU network and UW SCOR study. Reprod. Sci. 2008, 15, 866–877. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, Y.; Ren, S.; Liu, D.; Yang, N.; Han, Q.; Kong, S.; Wang, H.; Deng, W.; Qi, H.; et al. Single-cell RNA-seq revealed diverse cell types in the mouse placenta at mid-gestation. Exp. Cell Res. 2021, 405, 112715. [Google Scholar] [CrossRef]
- Monkley, S.J.; Delaney, S.J.; Pennisi, D.J.; Christiansen, J.H.; Wainwright, B.J. Targeted disruption of the Wnt2 gene results in placentation defects. Development 1996, 122, 3343–3353. [Google Scholar] [CrossRef] [PubMed]
- Parr, B.A.; Cornish, V.A.; Cybulsky, M.I.; McMahon, A.P. Wnt7b regulates placental development in mice. Dev. Biol. 2001, 237, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, E.C.M.; van Montfoort, A.P.A.; Dumoulin, J.C.M.; Evers, J.L.H. Epigenetics and the placenta. Hum. Reprod. Update 2011, 17, 397–417. [Google Scholar] [CrossRef]
- Serman, L.; Dodig, D. Impact of DNA methylation on trophoblast function. Clin. Epigenetics 2011, 3, 7. [Google Scholar] [CrossRef]
- Schuster, J.; Uzun, A.; Stablia, J.; Schorl, C.; Mori, M.; Padbury, J.F. Effect of prematurity on genome wide methylation in the placenta. BMC Med. Genet. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Dhar, G.A.; Saha, S.; Mitra, P.; Nag Chaudhuri, R. DNA methylation and regulation of gene expression: Guardian of our health. Nucleus 2021, 64, 259–270. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef]
- Suzuki, S.; Shaw, G.; Renfree, M.B. Identification of a novel antisense noncoding RNA, ALID, transcribed from the putative imprinting control region of marsupial IGF2R 06 Biological Sciences 0604 Genetics. Epigenetics Chromatin 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Makaroun, S.; Himes, K. Differential Methylation of Syncytin-1 and 2 Distinguishes Fetal Growth Restriction from Physiologic Small for Gestational Age. Am. J. Perinatol. Rep. 2018, 08, e18–e24. [Google Scholar] [CrossRef]
- Gao, Y.; He, Z.; Wang, Z.; Luo, Y.; Sun, H.; Zhou, Y.; Huang, L.; Li, M.; Fang, Q.; Jiang, S. Increased expression and altered methylation of HERVWE1 in the human placentas of smaller fetuses from monozygotic, dichorionic, discordant twins. PLoS ONE 2012, 7, e33503. [Google Scholar] [CrossRef] [PubMed]
- Ruebner, M.; Strissel, P.L.; Langbein, M.; Fahlbusch, F.; Wachter, D.L.; Faschingbauer, F.; Beckmann, M.W.; Strick, R. Impaired cell fusion and differentiation in placentae from patients with intrauterine growth restriction correlate with reduced levels of HERV envelope genes. J. Mol. Med. 2010, 88, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired Cytotrophoblast Cell–Cell Fusion Is Associated With Reduced Syncytin and Increased Apoptosis in Patients With Placental Dysfunction. Mol. Reprod. Dev. 2008, 75, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Kudaka, W.; Oda, T.; Jinno, Y.; Yoshimi, N.; Aoki, Y. Cellular Localization of Placenta-Specific Human Endogenous Retrovirus (HERV) Transcripts and their Possible Implication in Pregnancy-Induced Hypertension. Placenta 2008, 29, 282–289. [Google Scholar] [CrossRef]
- Ruebner, M.; Strissel, P.L.; Ekici, A.B.; Stiegler, E.; Dammer, U.; Goecke, T.W.; Faschingbauer, F.; Fahlbusch, F.B.; Beckmann, M.W.; Strick, R. Reduced Syncytin-1 Expression Levels in Placental Syndromes Correlates with Epigenetic Hypermethylation of the ERVW-1 Promoter Region. PLoS ONE 2013, 8, e56145. [Google Scholar] [CrossRef]
- Yu, Y.; He, J.; Hu, L.; Jiang, L.; Fang, L.; Yao, G.; Wang, S.; Yang, Q.; Guo, Y.; Liu, L.; et al. Placensin is a glucogenic hormone secreted by human placenta. EMBO Rep. 2020, 21, e49530. [Google Scholar] [CrossRef]
- Fowden, A.L.; Comline, R.S.; Silver, M. Insulin secretion and carbohydrate metabolism during pregnancy in the mare. Equine Vet. J. 1984, 16, 239–246. [Google Scholar] [CrossRef]
- George, L.A.; Staniar, W.B.; Cubitt, T.A.; Treiber, K.H.; Harris, P.A.; Geor, R.J. Evaluation of the effects of pregnancy on insulin sensitivity, insulin secretion, and glucose dynamics in Thoroughbred mares. Am. J. Vet. Res. 2011, 72, 666–674. [Google Scholar] [CrossRef]
- Poulet, M.; Sirois, J.; Boyé, K.; Uetani, N.; Hardy, S.; Daubon, T.; Dubrac, A.; Tremblay, M.L.; Bikfalvi, A. PRL-2 phosphatase is required for vascular morphogenesis and angiogenic signaling. Commun. Biol. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Li, L.; Shi, H.; Zhang, M.; Guo, X.; Tong, F.; Zhang, W.; Zhou, J.; Wang, H.; Yang, S. Upregulation of metastasis-associated PRL-3 initiates chordoma in zebrafish. Int. J. Oncol. 2016, 48, 1541–1552. [Google Scholar] [CrossRef]
- Guo, K.; Li, J.; Wang, H.; Osato, M.; Tang, J.P.; Quah, S.Y.; Gan, B.Q.; Zeng, Q. PRL-3 initiates tumor angiogenesis by recruiting endothelial cells in vitro and in vivo. Cancer Res. 2006, 66, 9625–9635. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; McQueeney, K.E.; Isenberg, J.S.; Pitt, B.R.; Wasserloos, K.A.; Homanics, G.E.; Lazo, J.S. Protein-tyrosine phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. J. Biol. Chem. 2014, 289, 5904–5913. [Google Scholar] [CrossRef] [PubMed]
- Croix, B.S.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montegomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Saha, S.; Sager, J.A.; Romans, K.E.; Xin, B.; Markowitz, S.D.; Lengauer, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B. PRL-3 Expression in Metastatic Cancers. Clin. Cancer Res. 2003, 9, 5607–5615. [Google Scholar] [PubMed]
- Haneda, S.; Dini, P.; Esteller-Vico, A.; Scoggin, K.E.; Squires, E.L.; Troedsson, M.H.; Daels, P.; Nambo, Y.; Ball, B.A. Estrogens regulate placental angiogenesis in horses. Int. J. Mol. Sci. 2021, 22, 12116. [Google Scholar] [CrossRef]
- Dini, P.; Carossino, M.; Balasuriya, U.B.R.; El-Sheikh Ali, H.; Loux, S.C.; Esteller-Vico, A.; Scoggin, K.E.; Loynachan, A.T.; Kalbfleisch, T.; De Spiegelaere, W.; et al. Paternally expressed retrotransposon Gag-like 1 gene, RTL1, is one of the crucial elements for placental angiogenesis in horses. Biol. Reprod. 2021, 104, 1386–1399. [Google Scholar] [CrossRef] [PubMed]
- Dini, P.; Carossino, M.; Loynachan, A.T.; El-Sheikh Ali, H.; Wolfsdorf, K.E.; Scoggin, K.E.; Daels, P.; Ball, B.A. Equine hydrallantois is associated with impaired angiogenesis in the placenta. Placenta 2020, 93, 101–112. [Google Scholar] [CrossRef]
- Tunster, S.J.; Creeth, H.D.J.; John, R.M. The imprinted Phlda2 gene modulates a major endocrine compartment of the placenta to regulate placental demands for maternal resources. Dev. Biol. 2016, 409, 251–260. [Google Scholar] [CrossRef]
- Frank, D.; Mendelsohn, C.L.; Ciccone, E.; Svensson, K.; Ohlsson, R.; Tycko, B. A novel pleckstrin homology-related gene family defined by lpl/Tssc3, TDAG51, and Tih1: Tissue-specific expression, chromosomal location, and parental imprinting. Mamm. Genome 1999, 10, 1150–1159. [Google Scholar] [CrossRef]
- Dunwoodie, S.L.; Beddington, R.S.P. The expression of the imprinted gene Ipl is restricted to extra-embryonic tissues and embryonic lateral mesoderm during early mouse development. Int. J. Dev. Biol. 2002, 46, 459–466. [Google Scholar]
- Takao, T.; Asanoma, K.; Tsunematsu, R.; Kato, K.; Wake, N. The maternally expressed gene Tssc3 regulates the expression of MASH2 transcription factor in mouse trophoblast stem cells through the AKT-Sp1 signaling pathway. J. Biol. Chem. 2012, 287, 42685–42694. [Google Scholar] [CrossRef] [PubMed]
- Tunster, S.J.; Tycko, B.; John, R.M. The Imprinted Phlda2 Gene Regulates Extraembryonic Energy Stores. Mol. Cell. Biol. 2010, 30, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; John, R.; Saxena, A.; Barton, S.; Frank, D.; Fitzpatrick, G.; Higgins, M.J.; Tycko, B. Placental growth retardation due to loss of imprinting of Phlda2. Mech. Dev. 2004, 121, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.B.; Tunster, S.J.; Heazell, A.E.P.; John, R.M. Placental PHLDA2 expression is increased in cases of fetal growth restriction following reduced fetal movements. BMC Med. Genet. 2016, 17, 1–5. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and Epigenetic Dynamics During Specification of Human Embryonic Stem Cells. Cell Stem Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef]
- Habibi, E.; Brinkman, A.B.; Arand, J.; Kroeze, L.I.; Kerstens, H.H.D.; Matarese, F.; Lepikhov, K.; Gut, M.; Brun-Heath, I.; Hubner, N.C.; et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell 2013, 13, 360–369. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Gim, J.A.; Hong, C.P.; Kim, D.S.; Moon, J.W.; Choi, Y.; Eo, J.; Kwon, Y.J.; Lee, J.R.; Jung, Y.D.; Bae, J.H.; et al. Genome-wide analysis of DNA methylation before- and after exercise in the thoroughbred horse with MeDIP-seq. Mol. Cells 2015, 38, 210–220. [Google Scholar] [CrossRef]
- Kanehisa, M.; Susumu, G. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.; Irizarry, R.; Kingsford, C. Salmon: Fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Chr | Log2FC- Expression | Log2FC- Methylation | Context | Region | Comparison | Agreement between DEG and DMR |
|---|---|---|---|---|---|---|---|---|
| ENSECAG00000023637 | ADAM33 | 22 | −1.07 | 0.89 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000009251 | ATXN1 | 20 | 1.11 | −0.83 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000021591 | B4GALNT1 | 6 | −1.51 | −0.87 | CG | Exon | 4M vs. 10M | No |
| ENSECAG00000008566 | CTSE | 5 | −2.63 | −2.79 | CHH | Exon | 4M vs. 6M | No |
| ENSECAG00000015010 | CYP4F124 | 21 | −2.69 | −0.65 | CG | Exon | 4M vs. 10M | No |
| ENSECAG00000015010 | CYP4F124 | 21 | −2.69 | −0.55 | CG | Exon | 4M vs. 6M | No |
| ENSECAG00000013832 | DES | 6 | −1.70 | 1.27 | CG | Exon | 6M vs. 10M | Yes |
| ENSECAG00000022980 | ENTPD8 | 25 | −3.15 | −1.29 | CG | Exon | 4M vs. 6M | No |
| ENSECAG00000020345 | ESPN | 2 | 1.11 | 1.32 | CG | Exon | 4M vs. 10M | No * |
| ENSECAG00000014851 | ILDR2 | 5 | 1.02 | −0.64 | CG | Exon | 4M vs. 10M | Yes |
| ENSECAG00000011659 | KIAA1549 | 4 | 1.12 | 1.64 | CG | Exon | 4M vs. 6M | No * |
| ENSECAG00000014702 | MEDAG | 17 | −1.47 | −2.08 | CG | Exon | 4M vs. 10M | No |
| ENSECAG00000014702 | MEDAG | 17 | −1.47 | −1.84 | CG | Exon | 4M vs. 6M | No |
| ENSECAG00000024536 | NFE2 | 6 | 1.19 | 0.90 | CG | Exon | 4M vs. 10M | No * |
| ENSECAG00000014030 | Novel gene | 3 | −1.79 | −2.74 | CG | Exon | 4M vs. 10M | No |
| ENSECAG00000029317 | AKR7A3 | PJAA01003681.1 | −1.97 | 1.68 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000016720 | OBSCN | 14 | −3.60 | −0.35 | CG | Exon | 4M vs. 10M | No |
| ENSECAG00000016720 | OBSCN | 14 | −3.60 | 0.48 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000013202 | PLIN1 | 1 | 4.01 | −2.73 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000017152 | RNF17 | 17 | −2.09 | 0.94 | CG | Exon | 4M vs. 10M | Yes |
| ENSECAG00000024853 | RSPO2 | 9 | −1.24 | 6.34 | CHH | Exon | 6M vs. 10M | Yes |
| ENSECAG00000019227 | SLC9A7 | X | 1.71 | 0.87 | CG | Exon | 4M vs. 10M | No * |
| ENSECAG00000019227 | SLC9A7 | X | 1.71 | 0.90 | CG | Exon | 4M vs. 6M | No * |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.13 | CG | Exon | 4M vs. 10M | Yes |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.37 | CG | Exon | 4M vs. 6M | Yes |
| ENSECAG00000020605 | TRIM2 | 2 | 1.10 | 1.16 | CG | Exon | 6M vs. 10M | No * |
| ENSECAG00000007169 | TTC22 | 2 | −1.50 | 0.45 | CG | Exon | 6M vs. 10M | Yes |
| ENSECAG00000039959 | ZDBF2 | 18 | 2.35 | −6.15 | CHH | Exon | 4M vs. 10M | Yes |
| ENSECAG00000007090 | ZFR2 | 7 | −1.73 | −0.68 | CG | Exon | 6M vs. 10M | No |
| ENSECAG00000000207 | ACTA1 | 1 | −2.89 | −1.33 | CG | Intron | 4M vs. 6M | No |
| ENSECAG00000023637 | ADAM33 | 22 | −1.07 | 0.89 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000008835 | ANKRD44 | 18 | 1.49 | −2.27 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000008835 | ANKRD44 | 18 | 1.61 | 2.10 | CG | Intron | 6M vs. 10M | No * |
| ENSECAG00000020314 | ANO1 | 12 | 1.90 | 0.56 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000020461 | BMPR1A | 1 | 1.49 | −2.77 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000009553 | CADM3 | 5 | 2.07 | 1.01 | CG | Intron | 6M vs. 10M | No * |
| ENSECAG00000010078 | CIT | 8 | 1.33 | 7.18 | CHH | Intron | 4M vs. 10M | No * |
| ENSECAG00000010078 | CIT | 8 | 1.33 | 5.01 | CHH | Intron | 4M vs. 6M | No * |
| ENSECAG00000016852 | CPT1A | 12 | −1.88 | 0.94 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000008566 | CTSE | 5 | −2.63 | −2.79 | CHH | Intron | 4M vs. 6M | No |
| ENSECAG00000015010 | CYP4F124 | 21 | −2.69 | −0.65 | CG | Intron | 4M vs. 10M | No |
| ENSECAG00000015010 | CYP4F124 | 21 | −2.69 | −0.55 | CG | Intron | 4M vs. 6M | No |
| ENSECAG00000020795 | DAGLA | 12 | 1.28 | 1.02 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000006857 | DCAF10 | 25 | 1.02 | 0.99 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000013582 | DHRS3 | 2 | −1.08 | 0.68 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000019565 | DLX5 | 4 | −1.39 | −1.58 | CG | Intron | 4M vs. 10M | No |
| ENSECAG00000023607 | DOCK5 | 2 | 1.37 | 0.68 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000020345 | ESPN | 2 | 1.11 | 1.32 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000000014 | GAS6 | 17 | −1.03 | −0.82 | CG | Intron | 4M vs. 6M | No |
| ENSECAG00000001312 | GPR146 | 13 | −1.18 | 0.74 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000014851 | ILDR2 | 5 | 1.02 | −0.64 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000014968 | JMY | 14 | 1.73 | 0.86 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000011659 | KIAA1549 | 4 | 1.12 | 1.64 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000039058 | KRT6C | 6 | −5.81 | −6.16 | CHH | Intron | 4M vs. 6M | No |
| ENSECAG00000020216 | KRT7 | 6 | −1.09 | −2.73 | CHH | Intron | 4M vs. 10M | No |
| ENSECAG00000000296 | LAMC3 | 25 | 1.18 | −4.45 | CHH | Intron | 4M vs. 6M | Yes |
| ENSECAG00000021630 | LGSN | 20 | −5.34 | 1.29 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000021583 | LMOD1 | 30 | −1.45 | 1.03 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000023118 | MCC | 14 | 1.26 | −0.76 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000024512 | TNFRSF10B | 2 | 1.26 | −4.21 | CHH | Intron | 4M vs. 10M | Yes |
| ENSECAG00000022376 | Novel gene | 18 | 1.52 | 1.66 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000022376 | Novel gene | 18 | 1.52 | 1.06 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000033604 | SLC7A4 | 8 | 2.72 | 1.01 | CG | Intron | 4M vs. 6M | No * |
| ENSECAG00000022376 | Novel gene | 18 | 1.09 | −1.47 | CG | Intron | 6M vs. 10M | Yes |
| ENSECAG00000018904 | NOXA1 | 25 | −1.77 | −1.29 | CG | Intron | 4M vs. 6M | No |
| ENSECAG00000016720 | OBSCN | 14 | −3.60 | −0.35 | CG | Intron | 4M vs. 10M | No |
| ENSECAG00000020485 | PCOLCE2 | 16 | −1.11 | −1.80 | CG | Intron | 6M vs. 10M | No |
| ENSECAG00000001688 | PCSK6 | 1 | −1.36 | 4.83 | CHH | Intron | 4M vs. 10M | Yes |
| ENSECAG00000001688 | PCSK6 | 1 | −1.36 | 3.39 | CHH | Intron | 4M vs. 6M | Yes |
| ENSECAG00000023890 | PRRX2 | 25 | −1.11 | 3.56 | CHH | Intron | 4M vs. 10M | Yes |
| ENSECAG00000023890 | PRRX2 | 25 | −1.11 | 3.37 | CHH | Intron | 4M vs. 6M | Yes |
| ENSECAG00000037450 | PTPRB | 6 | 1.13 | −0.61 | CG | Intron | 6M vs. 10M | Yes |
| ENSECAG00000026963 | PTPRR | 6 | −4.59 | 0.57 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000009250 | RAMP1 | 6 | −1.04 | 1.12 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000017152 | RNF17 | 17 | −2.09 | 0.94 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000020875 | RUNX2 | 20 | 1.55 | 0.84 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000019793 | SETBP1 | 8 | 1.70 | −1.03 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000009334 | SLC15A1 | 17 | −2.27 | 1.62 | CG | Intron | 6M vs. 10M | Yes |
| ENSECAG00000006302 | SLC25A29 | 24 | −1.29 | −1.22 | CG | Intron | 4M vs. 6M | No |
| ENSECAG00000014155 | SMOC2 | 31 | −1.62 | 1.48 | CG | Intron | 6M vs. 10M | Yes |
| ENSECAG00000022037 | SOX9 | 11 | −1.91 | 1.26 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000015256 | SPAG9 | 11 | 1.41 | 1.42 | CG | Intron | 4M vs. 10M | No * |
| ENSECAG00000008819 | TASOR2 | 29 | 1.05 | −0.94 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000008819 | TASOR2 | 29 | 1.05 | −1.01 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000008038 | TENM4 | 7 | 1.03 | 0.74 | CG | Intron | 6M vs. 10M | No * |
| ENSECAG00000007718 | TMOD1 | 25 | −2.01 | −6.62 | CHH | Intron | 6M vs. 10M | No |
| ENSECAG00000026887 | TSPAN8 | 6 | −1.54 | −0.96 | CG | Intron | 6M vs. 10M | No |
| ENSECAG00000013193 | UHRF1 | 7 | 1.37 | −2.17 | CG | Intron | 4M vs. 10M | Yes |
| ENSECAG00000013193 | UHRF1 | 7 | 1.37 | −2.19 | CG | Intron | 4M vs. 6M | Yes |
| ENSECAG00000022984 | ZFHX3 | 3 | 1.50 | 1.41 | CG | Intron | 6M vs. 10M | No * |
| ENSECAG00000007090 | ZFR2 | 7 | −1.73 | −0.68 | CG | Intron | 6M vs. 10M | No |
| ENSECAG00000024955 | ZFYVE28 | 3 | −5.29 | −2.03 | CG | Intron | 4M vs. 10M | No |
| ENSECAG00000016852 | CPT1A | 12 | −1.88 | 1.53 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000013582 | DHRS3 | 2 | −1.08 | 0.68 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000012011 | FBN2 | 14 | 1.43 | −1.13 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000012011 | FBN2 | 14 | 1.43 | −1.16 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000001312 | GPR146 | 13 | −1.18 | 0.87 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000016708 | HSF4 | 3 | −1.04 | 1.27 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000016708 | HSF4 | 3 | −1.04 | −1.45 | CG | Promoter | 6M vs. 10M | No |
| ENSECAG00000023496 | IRX3 | 3 | −5.44 | 1.57 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000021630 | LGSN | 20 | −5.34 | 1.29 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000013691 | MALT1 | 8 | 1.55 | −5.04 | CHH | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000033406 | Novel gene | 30 | 2.30 | 0.78 | CG | Promoter | 4M vs. 10M | No |
| ENSECAG00000029317 | AKR7A3 | PJAA01003681.1 | −1.97 | 1.68 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000000991 | PNMA3 | X | 1.68 | 1.41 | CG | Promoter | 4M vs. 6M | No |
| ENSECAG00000000484 | PPARA | 28 | 1.11 | −1.59 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000024853 | RSPO2 | 9 | −1.24 | 6.34 | CHH | Promoter | 6M vs. 10M | Yes |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.13 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.37 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000008819 | TASOR2 | 29 | 1.05 | −0.94 | CG | Promoter | 4M vs. 10M | Yes |
| ENSECAG00000008819 | TASOR2 | 29 | 1.05 | −1.01 | CG | Promoter | 4M vs. 6M | Yes |
| ENSECAG00000001072 | TYMP | 28 | −2.34 | −1.16 | CG | Promoter | 6M vs. 10M | No |
| ENSECAG00000001072 | TYMP | 28 | −2.34 | −1.27 | CG | Promoter | 6M vs. 10M | No |
| ENSECAG00000013202 | PLIN1 | 1 | 4.01 | −2.73 | CG | TES | 4M vs. 6M | Yes |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.13 | CG | TSS | 4M vs. 10M | Yes |
| ENSECAG00000001372 | SYN1 | X | 1.75 | −1.37 | CG | TSS | 4M vs. 6M | Yes |
| ENSECAG00000007090 | ZFR2 | 7 | −1.73 | −0.68 | CG | Utr3 | 6M vs. 10M | No |
| ENSECAG00000029317 | AKR7A3 | PJAA01003681.1 | −1.97 | 1.68 | CG | Utr5 | 4M vs. 6M | Yes |
| ENSECAG00000024853 | RSPO2 | 9 | −1.24 | 6.34 | CHH | Utr5 | 6M vs. 10M | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orellana-Guerrero, D.; Uribe-Salazar, J.M.; El-Sheikh Ali, H.; Scoggin, K.E.; Ball, B.; Daels, P.; Finno, C.J.; Dini, P. Dynamics of the Equine Placental DNA Methylome and Transcriptome from Mid- to Late Gestation. Int. J. Mol. Sci. 2023, 24, 7084. https://doi.org/10.3390/ijms24087084
Orellana-Guerrero D, Uribe-Salazar JM, El-Sheikh Ali H, Scoggin KE, Ball B, Daels P, Finno CJ, Dini P. Dynamics of the Equine Placental DNA Methylome and Transcriptome from Mid- to Late Gestation. International Journal of Molecular Sciences. 2023; 24(8):7084. https://doi.org/10.3390/ijms24087084
Chicago/Turabian StyleOrellana-Guerrero, Daniela, José M. Uribe-Salazar, Hossam El-Sheikh Ali, Kirsten E. Scoggin, Barry Ball, Peter Daels, Carrie J. Finno, and Pouya Dini. 2023. "Dynamics of the Equine Placental DNA Methylome and Transcriptome from Mid- to Late Gestation" International Journal of Molecular Sciences 24, no. 8: 7084. https://doi.org/10.3390/ijms24087084
APA StyleOrellana-Guerrero, D., Uribe-Salazar, J. M., El-Sheikh Ali, H., Scoggin, K. E., Ball, B., Daels, P., Finno, C. J., & Dini, P. (2023). Dynamics of the Equine Placental DNA Methylome and Transcriptome from Mid- to Late Gestation. International Journal of Molecular Sciences, 24(8), 7084. https://doi.org/10.3390/ijms24087084